


We present the case of a 45-year-old woman who was hospitalized due to severe macrocytic anemia and renal failure. The patient presented a morbid obesity.
The immunological study showed anti-ENA anti-SSA (Ro52) positive, with negative antinuclear antibodies. Also in the proteinogram (serum immunofixation) the presence of monoclonal bands IgG lambda and IgG kappa, monoclonal component 7.2% (4.68g/L), with elevation of free light chains (kappa 95.94mg/L (3.3–19.4), evidenced, lambda 145.17mg/L (5.71–26.3)).
The bone marrow study showed an infiltration of 5% of plasma cells and positive for AA amyloid. Finally, a percutaneous renal biopsy was performed, which again showed amyloid infiltration.
In the genetic study, 2 mutations of the family Mediterranean fever gene (MEFV) have been identified.
Secondary AA amyloidosis has been described associated with obesity, in addition to a percentage of cases of unknown etiology.
Presentamos el caso de una mujer de 45 años que fue hospitalizada debido a una anemia macrocítica severa e insuficiencia renal. El paciente presentaba una obesidad mórbida.
El estudio inmunológico mostró positividad para anti-ENA, anti-SSA (Ro52) y negatividad para anticuerpos antinucleares. También en el proteinograma (inmunofijación sérica) se detectó la presencia de bandas monoclonales IgG lambda e IgG kappa, con un componente monoclonal del 7,2% (4,68g/l) y la elevación de cadenas ligeras libres (kappa 95,94mg/l [3,3-19,4]; lambda 145,17mg/l [5,71-26,3]).
El estudio de biopsia de médula ósea mostró una infiltración del 5% de células plasmáticas y positividad para amiloide AA. Finalmente, se realizó una biopsia renal que nuevamente mostró infiltración amiloide.
En el estudio genético se identificaron 2 mutaciones del gen de la fiebre mediterránea familiar (MEFV).
La amiloidosis secundaria AA se ha descrito asociada a la obesidad, además de un porcentaje de casos de etiología desconocida.
Recent decades have led to the discovery of different amyloid types. Initially, amyloid was thought to represent a single entity. Are associated with clinically different diseases, including neoplasia and inflammatory, degenerative, genetic, and iatrogenic processes. Currently, 36 different protein, have been shown to be amyloidogenic.
An amyloid fibril protein is a protein that is deposited as insoluble fibrils, mainly in the extracellular spaces of organs and tissues as a result of sequential changes in protein folding that result in a condition known as amyloidosis.
An amyloid fibril protein occurs in tissue deposits as rigid, non-branching fibrils approximately 10nm in diameter. The fibrils bind the dye Congo red and exhibit green, yellow or orange birefringence when the stained deposits are viewed by polarization microscopy. When isolated from tissues and analyzed by X-ray diffraction, the fibrils exhibit a characteristic cross β diffraction pattern.1
The current amyloid nomenclature is based on the chemical structure of the fibril protein. Thus, a letter A (for amyloid) is followed by a suffix that is an abbreviated form of the precursor protein's name.
The form of amyloidosis AL currently represents about 80% of all forms of amyloidosis. The amyloidosis associated with an underlying plasma cell dyscrasia, when amyloid is derived from immunoglobulin light chains, the amyloid fibril is designated as AL and the disease is AL amyloidosis. In amyloidosis derived from the acute phase reactant serum amyloid A protein (SAA), the amyloid type is designated AA. Classifications of amyloid of different types based solely on clinical features, currently is not recommended. In hereditary amyloidoses that are associated with mutations in the amyloid protein, in addition to the general amyloid designation, the location of the mutation and the amino acid substitution they must be indicated.2,3 Amyloid deposits may be systemic or localized.
In the United States and the western world AL amyloidosis is the most common type of systemic amyloidosis, it constitutes 85% of all cases of systemic amyloidosis. The second most common type is AA amyloidosis. In case of AA amyloidosis, amyloid fibrils are derived from a truncated SAA, which is a major acute phase reactant. It is understood that SAA plays a role in inflammation and pathogen defense. Thus, AA amyloidosis develops in association with an enhanced and prolonged inflammation that leads to a sustained upregulated production of SAA and, subsequently, to its incomplete degradation, misfolding, and deposition in the tissues. In autoinflammatory diseases (including Crohn disease and familial Mediterranean fever) upregulated production of SAA is due to genetic defects in proteins involved in the modulation of the inflammatory response. This type amyloidosis affects the kidney and gastrointestinal tract. In case of nephritic syndrome, the most frequent clinical presentation is proteinuria.1
Due to the great diversity of fibrillar amyloid proteins that can be deposited, the possibility of different concomitant processes in patients with amyloidosis, and the various modern treatments for the different entities, in addition to the histopathological confirmation of congofilia in the deposits, it is mandatory to perform the typing of the fiber specifically deposited. To carry out chemical identification of an amyloid fibril protein, immunohistochemistry, Western blotting, mass spectrometry, after or without combination with laser capture, amino acid sequencing, and immune-electron microscopy are useful techniques.
The immunohistochemical technique, which uses specific monoclonal antibodies, is an effective method for the classification of amyloidosis that has shown a very high sensitivity and specificity of 100%.
We present the case of a patient with AA amyloidosis who has not been clinically associated with any chronic infectious, inflammatory or neoplastic disease.
Clinical caseA 45-year-old woman was hospitalized in January 2017 due to severe macrocytic anemia and renal failure. The patient presented a morbid obesity (height 155cm, weight 107.5, IMC 44.5), ex-smoker, without surgical interventions or known cardiopulmonary pathology. In the family history, he had a father and 2 grandparents who died of neoplasms (brain, liver and lung).
An analytical study was performed that showed a low reticulocyte number, severe folicopenia and vitamin B12 in the lower limit of normal, with alterations in the smear compatible with a deficiency origin. After being transfused on several occasions and folic acid supplements, B complex and erythropoietin have had an excellent response to treatment.
He also presented a positive Coombs in the context of ineffective erythropoiesis due to its megaloblastic anemia and very high beta 2 microglobulin (14.7mg/L) that was related to renal failure and ineffective erythropoiesis. A determination of occult blood in feces was also requested, which turned out to be negative.
On the other hand, at admission the patient had creatinine levels around 2.5mg/dl, with mild secondary metabolic acidosis and hyperkalemia, with 24-h proteinuria of 0.75g/day. In the urinary sediment, no hematuria or cylinders have been detected. Treatment with fluid therapy was instituted without improvement of renal function.
The patient, with anthropometric values of morbid obesity, presented parameters of analytical malnutrition at multiple levels: proteins, lipids, vitamins (particularly lipid soluble). Substitute treatment was performed, in addition to monitoring by endocrinology and nutrition during admission.
The patient recognized dietary restriction but not the eating behavior disorder, after assessment by mental health. There was no history of diarrhea/steatorrhea or abdominal pain, but there were data suggestive of a malabsorptive syndrome. They were requested stool studies that did not confirm maldigestion. The fecal calprotectin was slightly elevated, the anti-transglutaminase antibodies were negative and the endoscopic studies were not diagnostic.
Analytically it also showed very high levels of PTH (PTHi 574.9pg/mL) related to the marked deficit of vitamin D (D25 (OH) 6.3pg/mL) and chronic kidney disease. No alterations in blood and urine calcium levels were detected and phosphorus levels were normal. On the part of nephrology, studies were proposed to rule out primary hyperparathyroidism, performing a cervical ultrasound that was doubtful, not very suggestive and later a parathyroid scintigraphy that was normal.
During admission, after a weekend leave, the patient returned with pleuritic chest pain, dyspnea of minimal effort, productive cough, and leukocytosis, without fever. The examination revealed alterations in auscultation and a left basal alveolar infiltrate with a background interstitial pattern. The microbiological studies were negative (anti-core HBV, anti-HCV, anti-HIV, anti-CMV IgM, anti-Parvovirus B19 IgM, anti-EBV IgM VCA). The condition was interpreted as nosocomial pneumonia and empirical treatment with Piperacillin-Tazobactam and Clarithromycin was applied for 10 days, in addition to oxygen therapy. The inflammatory parameters were improving, disappearing the respiratory infectious clinic after the treatment. In the high endoscopic study, esophageal candidiasis attributable to the taking of antibiotics and steroids that he received temporarily was observed.
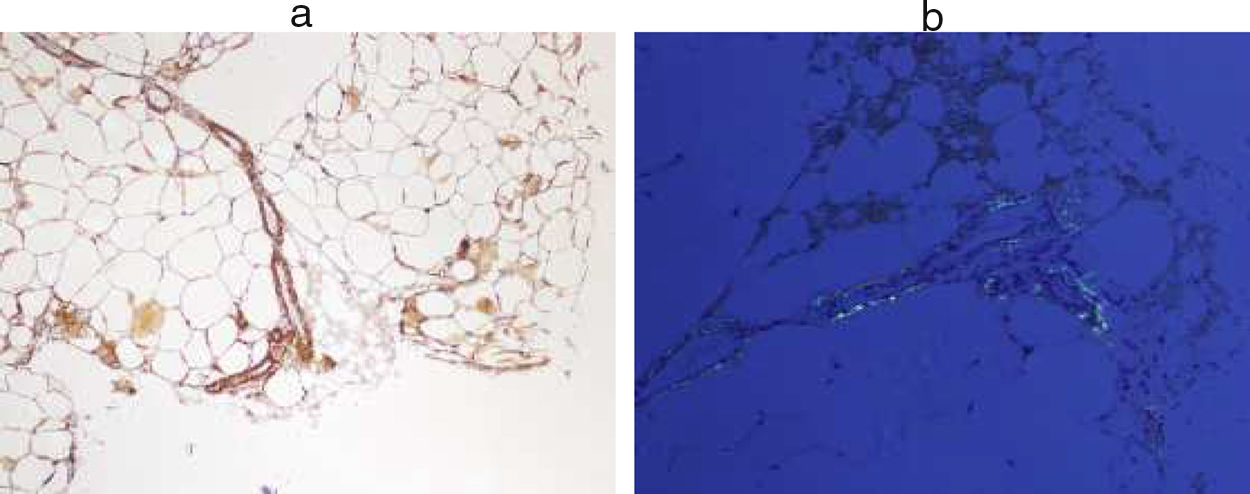
Immunological study showed anti-ENA anti-SSA (Ro52) positive, with negative antinuclear antibodies. Also in the proteinogram (serum immunofixation) the presence of monoclonal bands IgG lambda and IgG kappa, monoclonal component 7.2% (4.68g/L), with elevation of free light chains (kappa 95.94mg/L (3.3–19.4), was evidenced, lambda 145.17mg/L (5.71–26.3)). In the presence of anti-Ro antibodies positive and renal failure, despite not presenting symptoms suggestive of dry syndrome, studies were conducted to rule out Sjögren's syndrome. The use of a minor salivary gland biopsy was indicated to rule out focal lymphocytic sialadenitis. The result of FNA showed the presence of amyloid material. Subsequently, a FNA of abdominal fat was requested to typify the type of amyloid. In this sample the existence of AA amyloidosis was confirmed (Fig. 1).
 Amyloid AA. (b) Congo-red staining.")
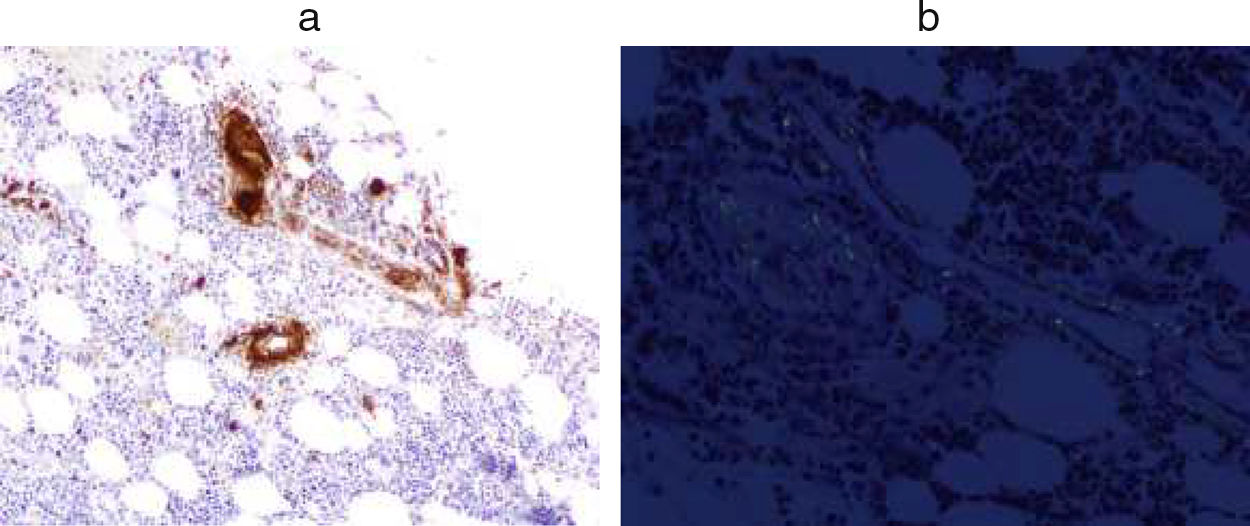
The study of bone marrow (Fig. 2) showed an infiltration of 5% of plasma cells and positive for Amyloid AA.
 Amyloid AA. (b) Congo-red.")
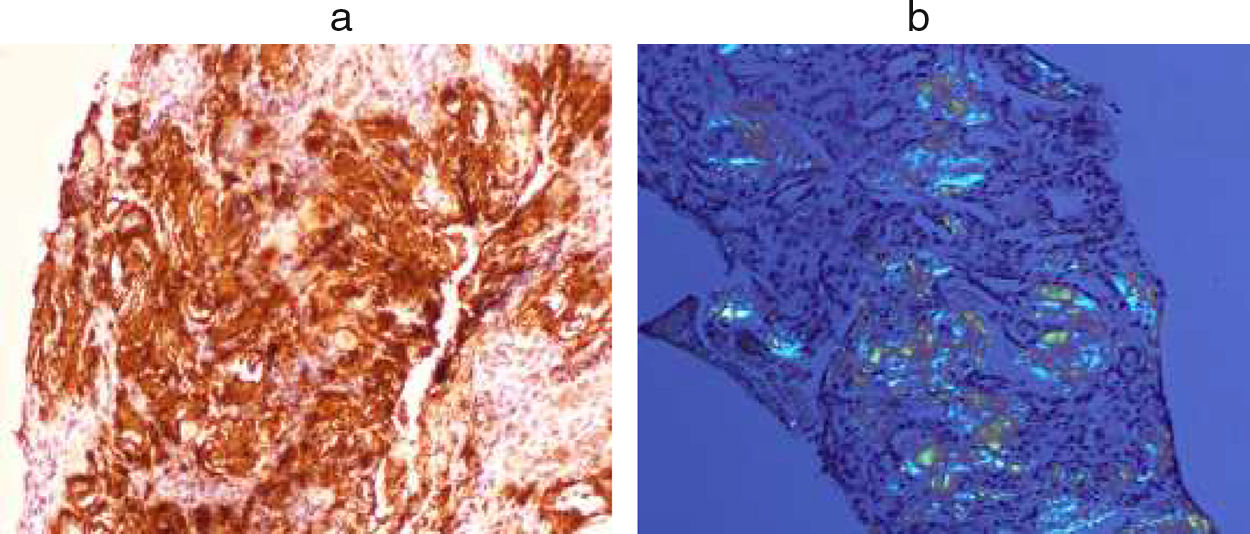
Finally, a percutaneous renal biopsy was performed, which again showed amyloid infiltration (Fig. 3).
 Amyloid AA. (b) Congo-red.")
The patient had no pathological history of interest to date; family history did not help clarify the etiology. During admission, multiple studies were performed that have allowed us to rule out infectious, inflammatory etiologies of another etiology, tumor or rheumatism.
Although from the clinical point of view it did not suggest the possibility of an autoinflammatory syndrome as a cause of its AA amyloidosis, genetic studies of periodic fevers were requested. In the genetic study, 2 mutations of the Familial Mediterranean Fever gene (MEFV) have been identified.
1. Presence in heterozygosis of the c.2230G>T (p.Ala744Ser) mutation in exon 10 of the MEFV gene.
2. Presence in heterozygosis of variant c.605G>A (p.Arg202Gln) in exon 2 of the MEFV gene.
The inhibitory treatment of IL-1 was established with anakinra, associating colchicine, evaluating the response and follow-up with the internal medicine service. In addition, reviews in outpatient nephrology and endocrinology.
DiscussionThe Mediterranean Family Fever has a pattern A: R (autosomal recessive). The c.2230G>T mutation (p.Ala744Ser) is described in the human gene mutation database (HGMD) and in the ClinVar, as associated with Familial Mediterranean Fever. And the variant c.605G>A (p.Arg202Gln) is described as an associated FMF polymorphism.
There is some controversy regarding the clinical significance of variant c.605G>A (p.Arg202Gln). According to some authors2,3 the presence of a pathogenic mutation (p.Ala744Ser) in heterosigosis together with the variant p.Arg202Gln is considered associated with Familial Mediterranean Fever. In this patient, this diagnosis could be considered, presenting a variant characterized by the existence of amyloidosis as the first clinical manifestation in an asymptomatic person.
FMF patients who develop amyloidosis generally have classic FMF with two inherited mutations. It is exceedingly rare, although not unheard of, for patients with single mutation FMF to develop amyloidosis. Such is the case with multiple members of a Spanish family with a H478Y mutation that causes an autosomal dominant form of FMF. AA amyloidosis has developed in many of the mutation positive members of this family.4
Secondary AA amyloidosis has been described associated with obesity,5 in addition to a percentage of cases of unknown etiology.
Some studies have shown that seric amyloid is expressed by subcutaneous white adipose tissue, and its production at this site is regulated by nutritional status. If amyloidosis is observed in the context of obesity, it is possible that the production of seric amyloid by adipocytes could be a contributing factor.6
There is emerging evidence that obese people carry an increased risk for the development of AA amyloidosis. Studies have shown that in obese people, SAA is produced by adipocytes, resulting in chronically elevated levels of SAA.6,7 Adipocytes treated with SAA release inflammatory cytokines as well as having decreased secretion of adiponectin.8 There is a report of a morbidly obese patient developing AA amyloidosis in the absence of any other known inflammatory condition. This report supports the theory that chronic production of SAA by adipocytes can contribute to the development of AA amyloidosis.9 Further investigation is needed to provide additional evidence of this observation.
The treatments are directed fundamentally toward the basic disease in case of being diagnosed.10–13
Conflict of interestThe authors declare no conflict of interest.


