


Las miopatías inflamatorias idiopáticas son un grupo heterogéneo de miopatías potencialmente tratables. Se clasifican en 4 subtipos: dermatomiositis, polimiositis, miositis autoinmune necrosante y miositis por cuerpos de inclusión, en función de las características clínicas e histológicas. Los anticuerpos asociados a miositis y los autoanticuerpos específicos de miositis se encuentran frecuentemente en pacientes con miopatías inflamatorias, siendo útiles en el diagnóstico y clasificación. El anticuerpo anti-histidil tRNA sintetasa es el más prevalente y el más específico para polimiositis. El anticuerpo de partícula de reconocimiento de señal es también un autoanticuerpo especıfico para polimiositis, pero más infrecuente, y raramente se encuentra en pacientes que presentan otros autoanticuerpos específicos para miositis.
En este trabajo se presenta un paciente con polimiositis en el que coexisten los 2 autoanticuerpos en el suero, lo que se considera una situación clínica extremadamente rara. Aquí analizamos la evolución clínica y hallazgos para examinar el efecto de la coexistencia y la posible interacción sobre el pronóstico.
Idiopathic inflammatory myopathies are a heterogeneous group of potentially treatable myopathies. They are classified, on the basis of clinical and histopathological features, into four subtypes: dermatomyositis, polymyositis, necrotizing autoimmune myositis and inclusion-body myositis. Myositis-associated antibodies and myositis-specific autoantibodies are frequently found in patients with idiopathic inflammatory myopathies, and are useful in the diagnosis and classification. Anti-histidyl transfer RNA synthetase antibody is the most widely prevalent and is highly specific for polymyositis. Signal recognition particle antibody is also a specific autoantibody for polymyositis, but it is infrequent and rarely found in patients having other myositis-specific autoantibodies.
We present a man with polymyositis who had both antibodies in serum, which is considered an extremely rare clinical situation. Here we analyze the clinical course and findings, and examine the effect of the coexistence and possible interaction on prognosis.
Las miopatías inflamatorias idiopáticas (MII) son miopatías potencialmente tratables, que se clasifican en: dermatomiositis, polimiositis (PM), miositis autoinmune necrosante y miositis por cuerpos de inclusión. En pacientes con MII son frecuentes los autoanticuerpos específicos de miositis, siendo útiles en el diagnóstico y clasificación. El anticuerpo anti-histidil-tRNA-sintetasa (anti-Jo1) es el más prevalente, y junto con el anti-partícula de reconocimiento de señal (anti-SRP) los más específicos para PM. Anti-SRP es infrecuente y aparece aislado en miositis.
Presentamos un caso clínico extremadamente raro, donde coexisten anti-Jo-1 y anti-SRP en el suero. Analizamos la evolución clínica para examinar el efecto de la coexistencia y posible interacción de ambos sobre el pronóstico.
Observación clínicaVarón de 60 años, fumador (40 paquetes/año), hipertenso y dislipidémico, tratado con atenolol 50mg/24h, ácido acetilsalicílico 100mg/24h, enalapril 10mg/24h, ezetimiba 10mg/24h y simvastatina 20mg/24h (esta desde septiembre de 2011 hasta febrero de 2012; se suspendió por el diagnóstico de PM).
Cuando el paciente fue estudiado presentó síndrome constitucional de 2 meses de evolución, con fiebre vespertina (38°C) que remitía con paracetamol; astenia, hiporexia y pérdida de peso (10kg). Refirió debilidad proximal, tos seca no productiva nocturna, artromialgias transitorias autolimitantes; no mostraba fenómeno de Raynaud ni trastornos dérmicos.
En la auscultación pulmonar se objetivaron estertores crepitantes finos en las bases y fuerte debilidad muscular axial y proximal de las extremidades.
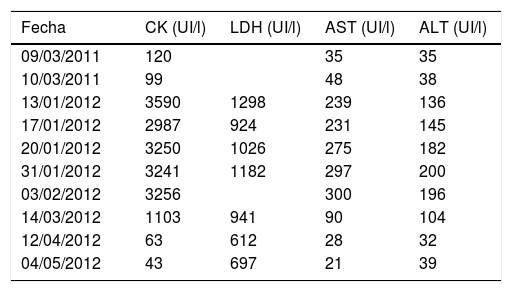
Las pruebas de laboratorio mostraron niveles séricos altos de creatín cinasa, lactato deshidrogenasa, alanino aminotransferasa, aspartato aminotransferasa (tabla 1), proteína C reactiva (79,2mg/l), velocidad de sedimentación globular (37mm/h) y leucocitosis (13.200U/mm3; 76,5% neutrófilos), con TSH normal (1,90μUI/ml). Solo los marcadores tumorales Ca 15.3 (54,8U/ml; normal: 2,0-37,0) y β2-microglobulina (4,63mg/l; normal≤3,0) estuvieron altos, aunque la ausencia de tumor se comprobó mediante tomografía computarizada de tórax y abdomen. El PPD fue negativo.
Evolución de los valores séricos de CK, LDH, AST y ALT
| Fecha | CK (UI/l) | LDH (UI/l) | AST (UI/l) | ALT (UI/l) |
|---|---|---|---|---|
| 09/03/2011 | 120 | 35 | 35 | |
| 10/03/2011 | 99 | 48 | 38 | |
| 13/01/2012 | 3590 | 1298 | 239 | 136 |
| 17/01/2012 | 2987 | 924 | 231 | 145 |
| 20/01/2012 | 3250 | 1026 | 275 | 182 |
| 31/01/2012 | 3241 | 1182 | 297 | 200 |
| 03/02/2012 | 3256 | 300 | 196 | |
| 14/03/2012 | 1103 | 941 | 90 | 104 |
| 12/04/2012 | 63 | 612 | 28 | 32 |
| 04/05/2012 | 43 | 697 | 21 | 39 |
ALT: alanín aminotransferasa; AST: aspartato aminotransferasa; CK: creatín cinasa; LDH: lactato deshidrogenasa.
Se detectaron anticuerpos anticelulares en el suero. El perfil de polimiositis por inmunoblot (anti-Mi2, anti-Ku, anti Pm/SCL, anti-Jo1, anti-PL12, anti-PI7, anti-SSA 52kDa, anti-EJ, anti-OJ y anti-SRP) fue positivo para anti-Jo-1 y anti-SRP.
Se analizaron los haplotipos para celiaquía; el paciente fue portador heterocigoto para el haplotipo DRB1*03:01-DQB1*02:01-DQA1*05:01, aunque los títulos de anticuerpos anti-transglutaminasa IgG e IgA fueron negativos.
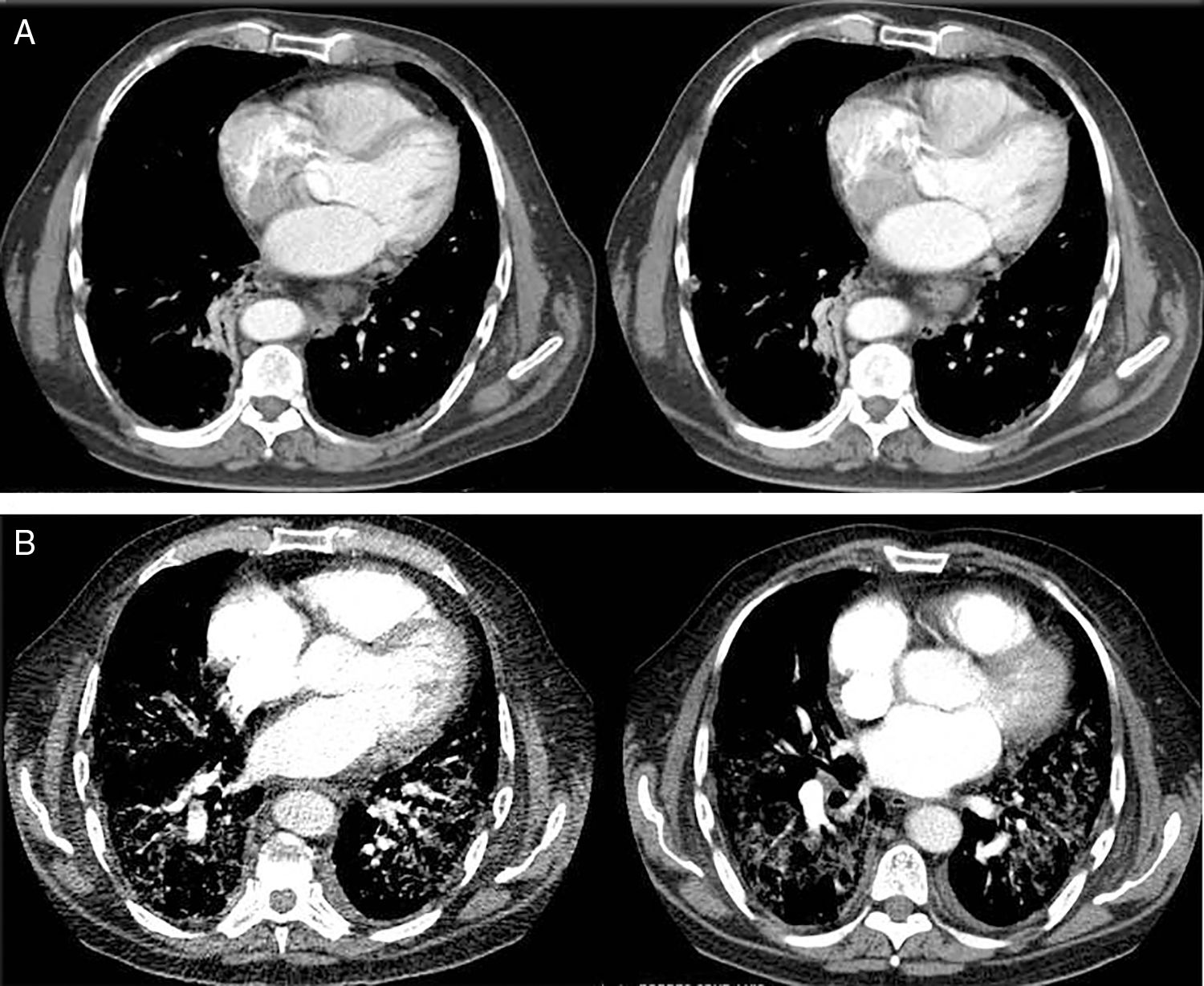
Los resultados de la tomografía computarizada se muestran en la figura 1, mientras que el estudio de broncoscopia mostró solo lesiones polipoideas supraglóticas. La citología de aspiración bronquial fue negativa para células neoplásicas.
. (A) La TAC de tórax al diagnóstico reveló la presencia de discretos infiltrados intersticiales en vidrio deslustrado en las bases y la periferia de los pulmones y, en menor medida, en los campos medios, con ganglios linfáticos mediastínicos múltiples de menos de 17mm y atelectasia segmentaria del lóbulo inferior derecho. (B) A los 3 meses la angioTAC no mostró imágenes de tromboembolismo pulmonar, pero sí se observaba una extensa afectación pulmonar bilateral con infiltrados intersticiales con opacidad en vidrio deslustrado y engrosamiento de los septos interlobulillares, así como áreas de fibrosis, estando más respetados los campos pulmonares anteriores.")
Evolución de la afectación pulmonar mediante tomografía axial computarizada (TAC). (A) La TAC de tórax al diagnóstico reveló la presencia de discretos infiltrados intersticiales en vidrio deslustrado en las bases y la periferia de los pulmones y, en menor medida, en los campos medios, con ganglios linfáticos mediastínicos múltiples de menos de 17mm y atelectasia segmentaria del lóbulo inferior derecho. (B) A los 3 meses la angioTAC no mostró imágenes de tromboembolismo pulmonar, pero sí se observaba una extensa afectación pulmonar bilateral con infiltrados intersticiales con opacidad en vidrio deslustrado y engrosamiento de los septos interlobulillares, así como áreas de fibrosis, estando más respetados los campos pulmonares anteriores.
El electromiograma mostró un proceso miopático predominantemente proximal, con presencia de signos discretos de actividad inflamatoria, que pueden aparecer en la polimiositis.
La biopsia de cuádriceps presentó discreta distorsión de la arquitectura fascicular con fibras perifasciculares atróficas ocasionales. Dentro de los fascículos se observaron fibras ocasionales con centralización nuclear y signos de regeneración. Destacaba la presencia de infiltrados inflamatorios linfomonocitarios perimisiales y endomisiales.
El diagnóstico se basó en las enzimas musculares elevadas, la biopsia muscular, los autoanticuerpos específicos de miositis y las anomalías del electromiograma.
Tras estos hallazgos se inició tratamiento oral con prednisona, 80mg/24h (1mg/kg/24h). El dolor muscular y la fiebre remitieron rápidamente, las enzimas musculares se normalizaron a los 3 meses (tabla 1). Sin embargo, el paciente presentó un episodio de trombosis venosa profunda y disnea progresiva, incluso en reposo, acompañada de tos no productiva. Se detectó cianosis en las áreas acrales, mostrando una saturación de O2 del 92% con máscara de oxigenoterapia con flujo alto.
La ecografía doppler venosa detectó un trombo oclusivo superficial en la vena femoral, en la vena poplítea y en el origen del tronco tibioperoneo. Con la terapia anticoagulante, 3 pulsos (1g/24h) por vía intravenosa de metilprednisolona y ciclofosfamida por vía intravenosa se consiguió una mejoría clínica.
DiscusiónLos anticuerpos anti-Jo-1 son los más frecuentes en MII (20-30%)1. Se asocian al síndrome anti-sintetasa, caracterizado por polimiositis, enfermedad intersticial pulmonar difusa, poliartritis, fenómeno de Raynaud y lesiones hiperqueratósicas de la piel, con eritema sobre las articulaciones metacarpofalángicas e interfalángicas2. Los pacientes con anti-Jo1 presentan miopatía (90%) y enfermedad intersticial difusa pulmonar (70%)3, pudiendo correlacionarse el título con la actividad de la enfermedad4.
Los anticuerpos anti-SRP reconocen la proteína citoplasmática que se une a las secuencias señal de proteínas sintetizadas para su translocación al retículo endoplásmico. Se encuentran en el 4% de adultos con polimiositis y no suelen asociarse a otros autoanticuerpos de miositis5. Pueden provocar la aparición aguda de polimiositis, con debilidad muscular grave, afectación cardíaca, progresión rápida, mala respuesta inmunosupresora y aumento de mortalidad6. Las biopsias musculares muestran abundantes signos de degeneración celular y regeneración, células necróticas y un discreto infiltrado inflamatorio. Sin embargo, los hallazgos de la biopsia de nuestro paciente no muestran este fenotipo anatomopatológico anti-SRP.
Clínicamente, los anticuerpos anti-Jo-1 se asocian con enfermedad intersticial pulmonar difusa y los anticuerpos anti-SRP con el inicio agudo de la enfermedad, el aumento sostenido de la creatín cinasa y la debilidad grave del paciente7, lo cual se manifestó en este paciente.
La forma paraneoplásica de miopatías inflamatorias es conocida, pero el tumor ha sido clínicamente descartado en nuestro paciente.
La toxicidad de las estatinas, presente en 7-29% de los casos, se asocia con miopatías inflamatorias8, además del hallazgo sérico del anticuerpo anti-3-hidroxi-3-metilglutaril-enzima-reductasa (anti-HMGCR), objetivo farmacológico de las estatinas. Los síntomas miopáticos suelen desaparecer al interrumpir el tratamiento, requiriéndose terapia inmunosupresora9. En el caso de la miopatía necrosante inducida por estatinas se ha visto la persistencia de los síntomas, incluso interrumpiendo el tratamiento, como ocurrió en nuestro paciente.
Sin embargo, en la biopsia muscular no se apreciaron fibras necróticas y regenerativas, pero sí focos de linfocitos, reforzando el diagnóstico de PM.
El haplotipo de celiaquía de este paciente confiere riesgo de miopatía autoinmune (razón de momios 3,6) y 15,5 veces riesgo de anti-Jo-110.
ConclusionesEl caso presentado aquí es inusual en la práctica clínica. La PM es una enfermedad de baja incidencia en España (3,9/100.000). La coexistencia de ambos autoanticuerpos se considera extremadamente rara, y en este caso además los anticuerpos fueron útiles para diagnosticar la miopatía autoinmune y el síndrome anti-sintetasa. La asociación con anticuerpos anti-SRP provocó un deterioro muscular grave y mal pronóstico.
Conflicto de interesesTodos los autores declaran no tener ningún conflicto de intereses en este manuscrito.


