


La enfermedad de Gaucher (EG) descrita en 1882 por Philippe Charles Ernest Gaucher es una enfermedad hereditaria poco frecuente, progresiva, con un patrón de herencia autosómico recesivo1. Incluida en el grupo de enfermedades por depósito lisosomal, se caracteriza por ser la más prevalente con una frecuencia estimada de 1/50.000 a 1/100.000 habitantes en la población general, a excepción de la etnia judía ashkenazí estimándose en 1/850 nacimientos2,3. Causa una deficiencia de la actividad de la enzima beta glucosidasa ácida (GBA), provocando acúmulo de glucocerebrósidos en los lisosomas de diferentes células4, causando citopenias, hepatoesplenomegalia, alteraciones del sistema nervioso central (SNC) y manifestaciones esqueléticas, constituyendo estas últimas uno de los aspectos más discapacitantes. Según la expresividad clínica se pueden distinguir diversos tipos: tipo 1 (no neuronopática adulta), la más común, frecuente en judíos de origen ashkenazí, con manifestaciones variables, sin afectación del SNC; tipo 2 (neuronopática aguda), infrecuente, sin dominio étnico, letal tras el nacimiento y afectación del SNC; tipo 3 (neuronopática subaguda o crónica)4, en la infancia, adolescencia o adultos, con afectación del SNC. Dada la variedad de entidades asociadas a dolor óseo, nos parece oportuno aportar el caso de nuestro paciente.
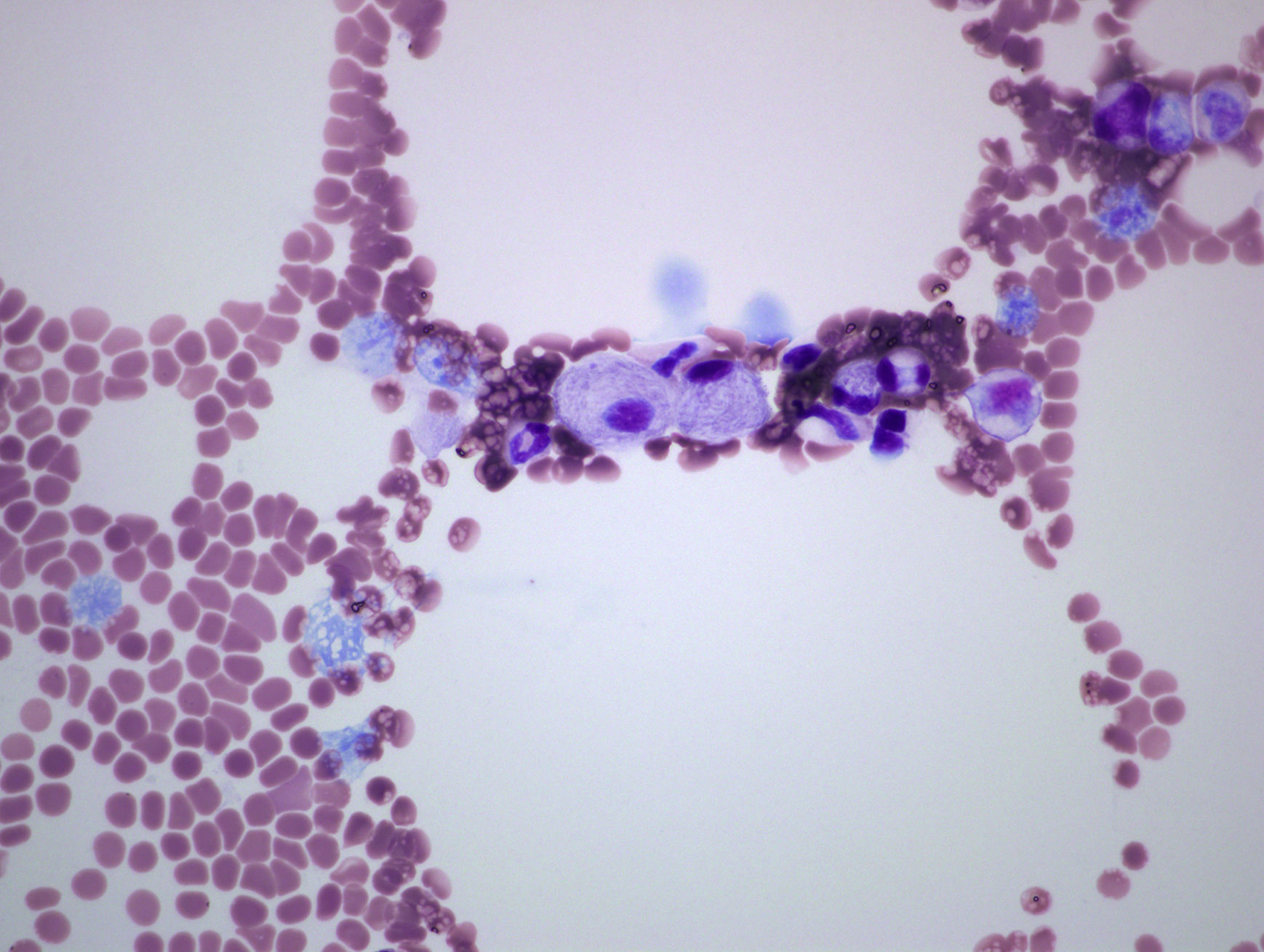
Varón de 53 años, sin antecedentes familiares ni personales de interés, valorado por neutropenia y trombocitopenia de 10 años de evolución y dolores óseos desde hacía 3 años. No refería diátesis hemorrágica, infecciones ni dolor abdominal. En la exploración destacó una esplenomegalia, siendo el resto normal. Los estudios de laboratorio mostraron leucocitos 3.400mm3, neutrófilos 1.200/mm3 y plaquetas 93.000mm3, el resto incluyendo hemoglobina, reticulocitos, coagulación, ionograma, función hepática, vitamina B12, ácido fólico, marcadores tumorales (antígeno carcinoembrionario, alfafetoproteína, CA 19-9, CA 15-3, antígeno prostático específico), ß2-microglobulina, factor reumatoide, VSG, ANA, inmunoglobulinas, proteinograma y poblaciones linfocitarias fueron normales. Las serologías virales para hepatitis B, C e inmunodeficiencia humana fueron negativas. La radiología simple de fémur distal y columna dorso-lumbar fueron normales, sin embargo, la resonancia magnética lumbar mostró hiposeñal homogénea en los cuerpos vertebrales en secuencia T1 y T2. La esplenomegalia fue confirmada con ecografía abdominal, y la osteoporosis con densitometría ósea (T-score fémur: −2,9 DE: T-score columna: −2,8 DE). El aspirado/biopsia de médula ósea (AMO/BMO), detectó células de núcleo excéntrico, citoplasma basófilo en papel de seda sugestivas de células de Gaucher (CG) (fig. 1). Se cuantificó la actividad enzimática GBA en leucocitos mediante espectrofluorimetría, confirmando déficit de la misma. El estudio genético molecular mostró doble heterocigosidad para las mutaciones L444P y p.Tyr244Cys.

La presente observación constituye un ejemplo representativo de las características clínicas, bioquímicas y genéticas de la EG tipo 15. El hecho de que nuestro paciente presente dolor óseo de años de evolución, puede dificultar la orientación diagnóstica, estando estas manifestaciones asociadas a otros datos de la enfermedad como son citopenias y visceromegalias, siendo las manifestaciones extra-articulares útiles para realizar un correcto diagnóstico, evitando falsos diagnósticos de enfermedades inflamatorias y/o autoinmunitarias. El AMO puso de manifiesto CG, cuya acumulación genera sustancias responsables de la resorción ósea, produciendo dolor, deformidad e incapacidad funcional6. Se puede detectar mediante exploraciones radiológicas manifestaciones como son alteración en la remodelación del contorno óseo (mostrando la deformidad en matraz de Erlenmeyer, que en nuestro caso no se encontró), fracturas espontáneas, osteopenia, osteonecrosis y osteólisis. Sin embargo, el desarrollo de osteoporosis de causa desconocida, asociada o no a trombocitopenia y esplenomegalia debe hacernos sospechar EG. Estos hallazgos fueron clave, por lo que se cuantificó la actividad de la enzima GBA y se solicitó un estudio genético para confirmar la enfermedad6. Se requiere alta sospecha y estudios bioquímicos iniciales que permitan llegar al diagnóstico, e iniciar terapia de reemplazo enzimático para revertir, estabilizar y mejorar los aspectos clínicos del paciente.


