


El síndrome de Muckle-Wells es una enfermedad autoinflamatoria sistémica incluida dentro del grupo de fiebres periódicas hereditarias.
Presentamos a un paciente con esta enfermedad para llamar la atención sobre la singularidad de esta entidad, su baja frecuencia y presentación atípica, que conllevan generalmente demora en el diagnóstico, cuando ya hay consecuencias tardías y muchas veces devastadoras.
En este caso, la terapia de primera línea antiinterleucina 1 (IL-1) no consiguió frenar la enfermedad, consiguiendo, sin embargo, su control el inhibidor de IL-6 tocilizumab, mostrándose eficaz en la remisión total del síndrome nefrótico asociado a amiloidosis secundaria AA, cambiando su oscuro pronóstico.
Muckle-Wells syndrome is a systemic autoinflammatory disease included in the group of hereditary periodic febrile syndromes.
We report the case of a patient with this rare disease to call the attention to the singularity of this condition, its low incidence, its atypical presentation and the subsequent delay in the diagnosis, which is reached when late and devastating consequences have taken place.
In this case, the first-line therapy, anti-interleukin 1 (IL-1), failed to control the disease. Nevertheless, the IL-6 inhibitor, tocilizumab, proved effective, achieving the total remission of nephrotic syndrome associated with AA secondary amyloidosis, changing the bleak prognosis of this disease.
El síndrome de Muckle-Wells es una enfermedad rara y hereditaria con un patrón de transmisión autosómico dominante. Está causado por la mutación del gen NLRP3 (también llamado CIAS1), que codifica la criopirina, proteína responsable de la regulación de la producción de citocinas inflamatorias, principalmente interleucina (IL)-1beta, lo que conlleva una inflamación sistémica persistente e incontrolada1. Existen 3 tipos de síndrome asociado a criopirinas (CAPS) de gravedad clínica creciente: síndrome autoinflamatorio familiar inducido por frío, síndrome de Muckle-Wells y síndrome CINCA-NOMID.
No existen unos criterios diagnósticos claramente definidos, pero según la literatura médica su diagnóstico se fundamenta en 3 pilares. En primer lugar, la clínica se inicia en forma de episodios caracterizados por fiebre, rash/urticaria y artralgias/artritis asociadas a dolor abdominal y conjuntivitis/uveítis anterior. Estos episodios son autolimitados y recurrentes, de 2 a 5 días de duración. De forma más tardía, se pueden encontrar una sordera neurosensorial progresiva y/o una amiloidosis secundaria AA (fundamentalmente con afectación renal); la primera presente en aproximadamente el 60% de los pacientes, mientras que la segunda en un 25%2.
El tratamiento se basa en primer lugar en antiinflamatorios no esteroideos (AINE), colchicina y corticoides, además de las terapias anti-IL-13 (principalmente anakinra, rilonacept y canakinumab)4.
Caso clínicoVarón de 25 años, que refería episodios recurrentes de fiebre prolongada desde los 2 años, con una periodicidad de 1-2 brotes anuales. En una revisión pediátrica se detectó la presencia de hepatoesplenomegalia. También presentaba artromialgias sin signos inflamatorios, algún episodio de dolor abdominal y en 2 ocasiones rash evanescente. En la familia no había antecedentes de fiebres recurrentes ni de colagenopatías. Fue valorado por los servicios de Pediatría, Reumatología, Hematología y Medicina Interna, con un primer diagnóstico de enfermedad de Still de comienzo sistémico, que fue tratado con AINE y paracetamol.
En la analítica presentaba de forma mantenida: leucocitosis, elevación de los reactantes de fase aguda, proteína C reactiva y velocidad de sedimentación globular. Sin embargo, los valores de ferritina, factor reumatoide, anticuerpos antinucleares (ANA), sistema del complemento e inmunoglobulinas fueron siempre negativos o dentro de la normalidad.
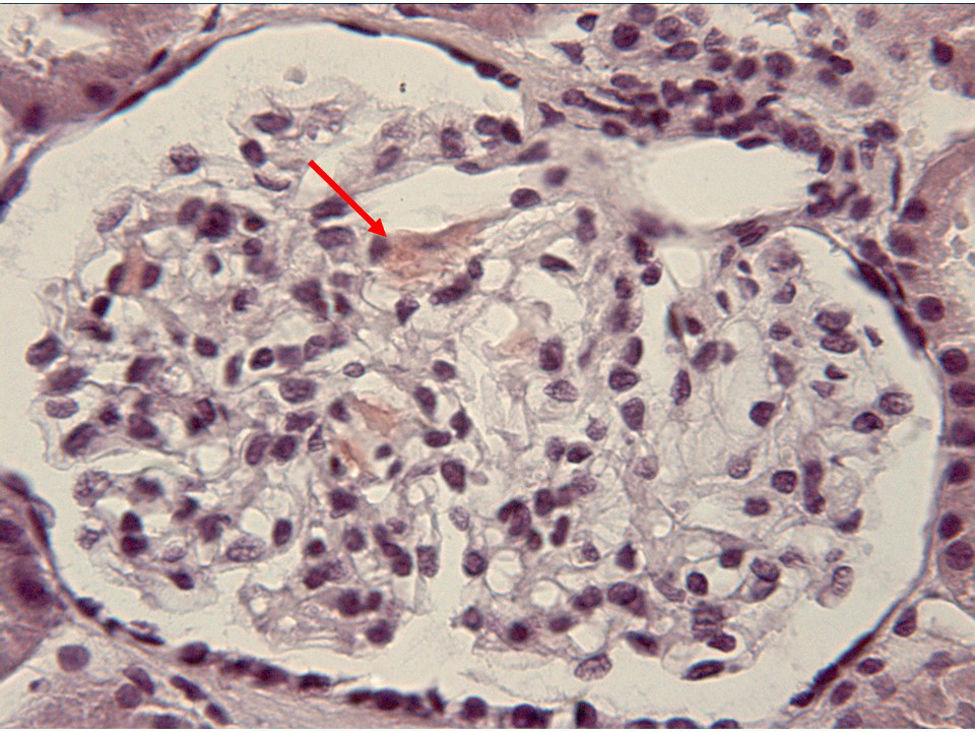
A los 23 años, se detectó la presencia de proteinuria en rango nefrótico sin alteraciones del sedimento. Ante la sospecha de una proteinuria en relación con la toma de AINE o con su proceso inflamatorio de base, se realizó una biopsia renal que mostró una amiloidosis secundaria (fig. 1). En ese momento, con la clínica y la analítica descritas, y tras la aparición de la amiloidosis renal, se replanteó el diagnóstico. Se pensó en la posibilidad de que se tratara de una enfermedad autoinflamatoria sistémica hereditaria más que de una enfermedad de Still y se solicitaron los estudios genéticos al respecto, con resultado positivo para la mutación del gen NLRP3, diagnosticándose entonces de síndrome de Muckle-Wells.
 del glomérulo del paciente, mostrando positividad para el amiloide depositado en el mesangio (flecha).")
Se inició entonces tratamiento con anakinra 100mg subcutáneos diarios4, sin conseguir un adecuado control de la proteinuria. Dada la mala respuesta y tras el hallazgo de unos niveles elevados de IL-6, se suspendió el anakinra y se inició tratamiento con tocilizumab (inhibidor de la IL-6). En los últimos 6 años, el paciente se ha mantenido asintomático, con buen control de la inflamación y con una función renal normal sin proteinuria.
DiscusiónEste paciente presentaba desde la infancia episodios recurrentes de fiebre y artralgias y, además, episodios de dolor abdominal y rash cutáneo. El factor reumatoide y los ANA fueron negativos, siendo todo ello compatible tanto con la enfermedad de Still5 como con las criopirinopatías. La coexistencia de hepatoesplenomegalia, según los criterios de Yamaguchi6, indicó el diagnóstico de enfermedad de Still. No fue hasta la aparición de la amiloidosis renal, 20 años después, cuando se replanteó el diagnóstico. El estudio genético confirmó el diagnóstico de síndrome de Muckle-Wells.
ConclusiónDada la rareza de estos síndromes en la práctica clínica habitual, hay que recalcar la importancia de conocer sus diferentes y a veces incompletas formas de presentación y mantener un alto nivel de sospecha para así poder plantear un adecuado diagnóstico diferencial, orientar las pruebas genéticas que confirmen el diagnóstico definitivo, e instaurar una terapia biológica adecuada. De esta manera, se conseguirá frenar, o al menos enlentecer, el avance de la enfermedad y con ello la aparición de complicaciones —amiloidosis renal— que puedan ensombrecer su pronóstico, como ocurrió en este caso. No obstante, el inhibidor de IL-6, tocilizumab, mostró eficacia en la remisión total del síndrome nefrótico, cambiando el pronóstico de esta enfermedad.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


