


Nephrogenic systemic fibrosis (NSF) is a fibrosing skin condition of unknown origin. Most cases have been described in patients with acute or chronic renal failure. The cutaneous changes include firm and thickened, indurate skin plaques and papules on the extremities and trunk. Histopathology typically shows an increase in dermal fibroblast-like cells associated with mucin deposition. Previous exposition to gadolinium-based contrast agents was closely associated with its onset. We described a patient with the clinical and pathologic picture of NSF presented after an acute renal failure in the course of a perinuclear antineutrophil cytoplasmic antibodies associated systemic vasculitis.
La fibrosis sistémica nefrogénica (FSN) es una entidad de origen desconocido caracterizada por un incremento de la fibrosis cutánea. La mayoría de los casos se han descrito en pacientes con fracaso renal agudo o crónico. Las alteraciones cutáneas incluyen pápulas y placas dermicas engrosadas e induradas distribuidas por las extremidades y el tronco. El estudio histológico pone de manifiesto un incremento en la dermis del número de células tipo fibroblasto acompañado de depósito de mucina. En la mayoría de los casos existe el antecedente reciente de exposición a agentes de contraste tipo gadolinio. Describimos una paciente que presenta los hallazgos clínicos y patológicos característicos de la FSN después de un fracaso renal agudo en el contexto de una vasculitis asociada a anticuerpos anticitoplasma de los neutrófilos.
Nephrogenic systemic fibrosis (NSF) is a newly fibrosing skin condition first identified in 1997, and the initial published report of 15 cases appeared in 20001. Most cases are described in patients with renal insufficiency undergoing haemodialysis, peritoneal dialysis or after cadaveric renal transplant. Some cases have been described in patients with acute renal failure (ARF). Numerous reports have been published, some of them in Europe2–9. The cutaneous changes include firm and thickened, indurate skin plaques and papules as well as nodules on the extremities and trunk. Histopathology typically shows an increase in dermal fibroblast-like cells associated with mucin deposition. Differential diagnosis includes scleromyxoedema, systemic or localized sclerodermia, eosinophilic fascitis, eosinophilia-myalgia syndrome, scleredema, lipodermatosclerosis, porphyria cutanea tarda, rheumatism fibroblastic, Spanish toxic oil syndrome, vinyl chloride exposure, dermatofibrosarcoma protuberans and β-2 microglobulin amyloidosis10. In 2006 Grobner and Marckmann proposed that Gadolinium based contrast agents (Gd-CA) may trigger the development of NFS in patients with underlying metabolic acidosis11,12. We described a patient with the clinical and pathologic picture of NSF presented after an ARF in the course of a perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) associated systemic vasculitis.
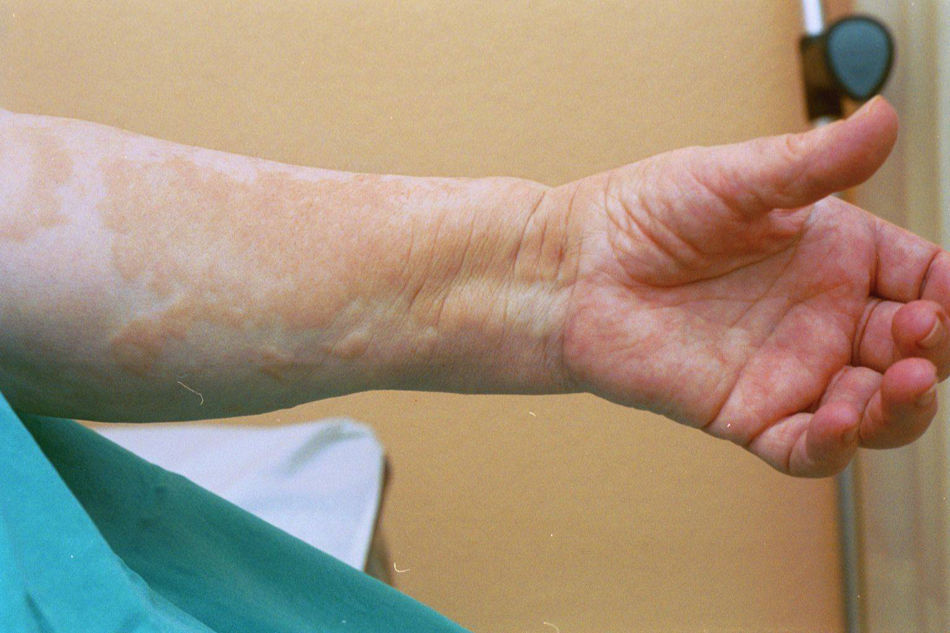
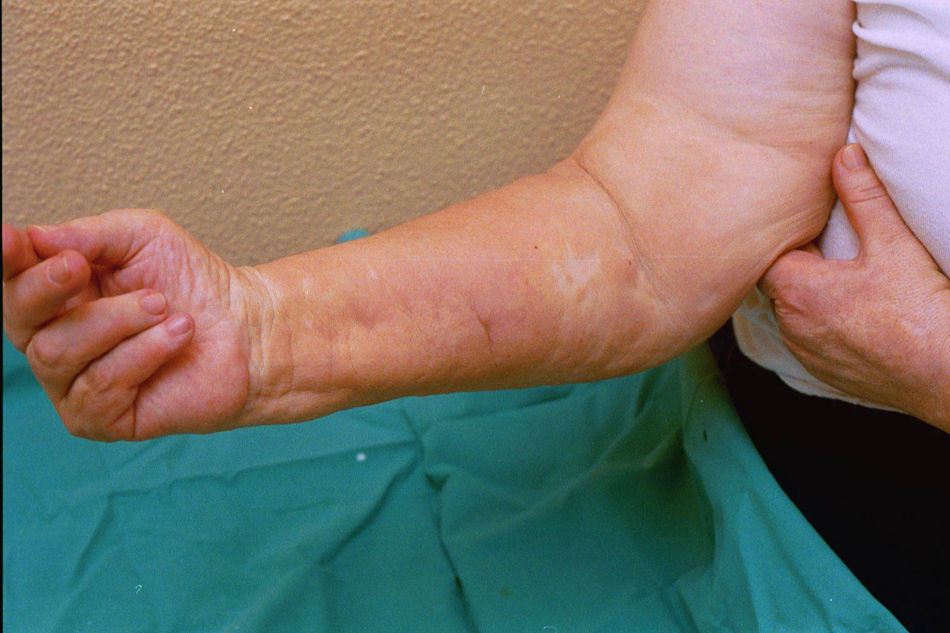
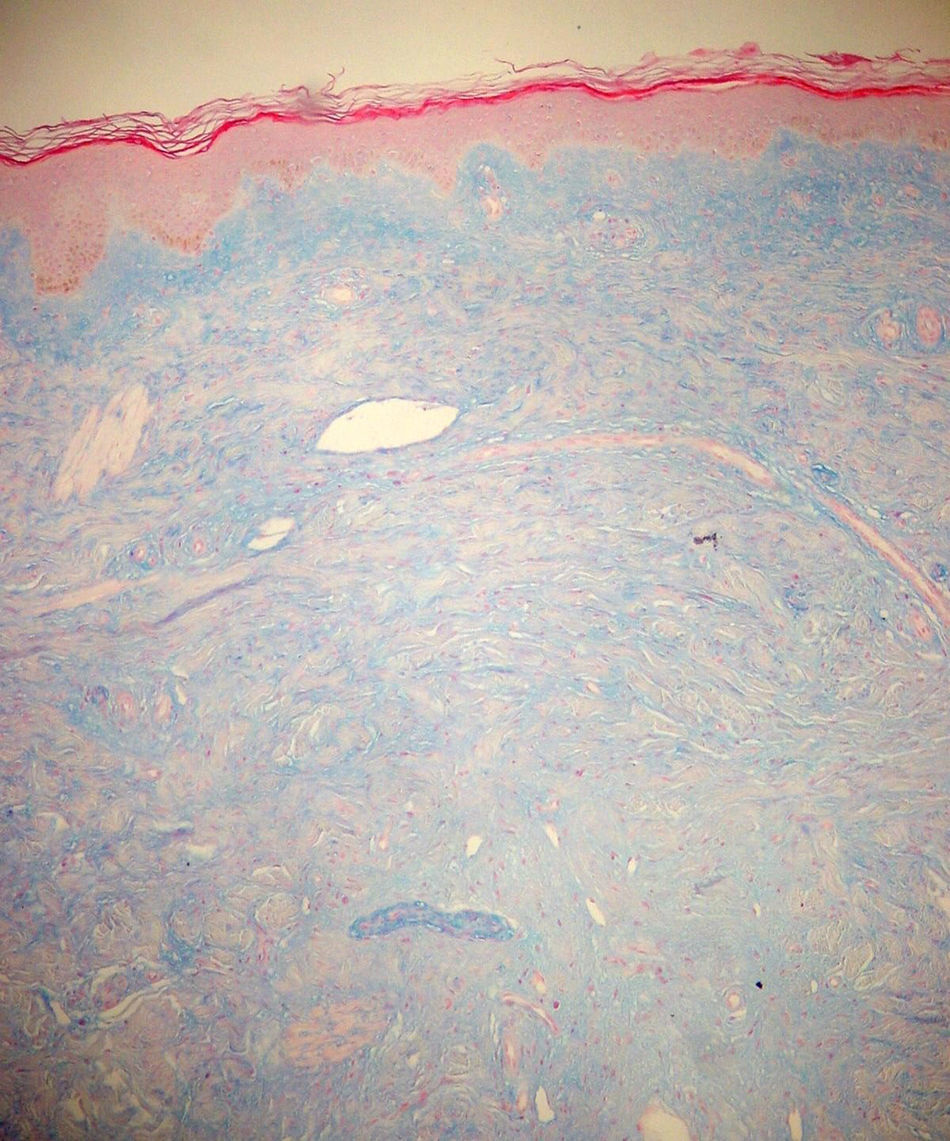
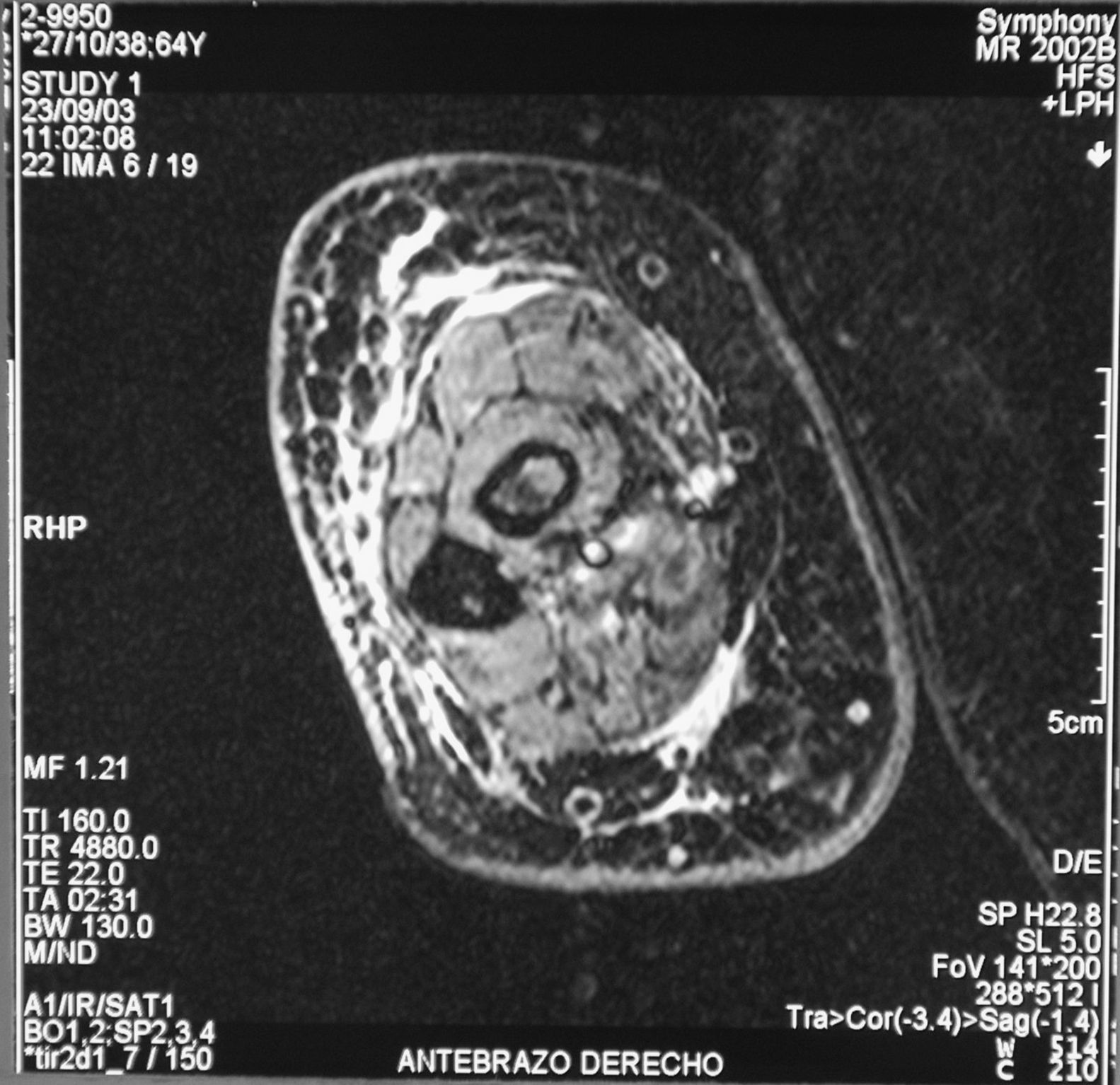
Case reportThe patient, a 65-year-old woman was referred to our institution in February 2003. Her medical history included pulmonary tuberculosis at 28 years of age, severe obesity and varicosity and phlebitic disease. The patient had been treated previously with alternative therapies. The family antecedents included a 39-year-old daughter suffering a rheumatoid arthritis and systemic lupus erythematosus overlap syndrome and another 38 year-old daughter diagnosed with a microscopic polyarteritis with positive ANA and p-ANCA that evolved to chronic renal failure requiring haemodialysis. In February 2003 the patient was admitted to another hospital because of a 4-week syndrome with fever, cough, dysnea and myalgias. Three days before being admitted the patient complained of pain, oedema and swelling of the right leg and increased dyspnoea and vomiting together with abdominal pain. On admittance acute renal failure with a creatinine of 2,33mg/dL, a normochromic normocytic anaemia (Hb 9.1mg/dl), an erythrocyte sedimentation rate (ESR) of 128 and a C-reactive protein of 21,7mg/L was reported. Arterial blood gases on air revealed a pa O2 of 60, a p CO2 of 35 and an O2 saturation of 94%. A chest radiography showed bilateral alveolar infiltrates first interpreted as infection. An echography of the right leg showed deep venous thrombosis of the internal saphena. At that point the patient did not accept the performance of a thoracic computed tomography (CT). Treatment with antibiotics, low weight heparin, and corticoids was instituted but the patient presented a gastric haemorrhage and a haematoma on the right leg and worsening of the anaemia that required blood transfusion and was referred to our hospital. On admittance the laboratory studies revealed an ESR of 13, normochromic normocytic anaemia (Hb 7.8mg/dl) with 22500 leucocytes, and platelet count and coagulation tests within normal range. The serum creatinine had increased to 2,8mg/dL. A urinalysis revealed 3+ leucocytes and 3+ blood dipstick but protein or casts were absent. The rheumatoid factor and cryoglobulins were negative. Antinuclear antibodies were positive to 1/640 by indirect immunofluorescence (Hep-2 cells) Anti-DNA and anticardiolipin antibodies were negative (including anti-β-2 glicoprotein I) C3 was 59,5mg/dl and C4 within range. Serologic tests for hepatitis B and C were negative. Anti-SS-A (Ro) antibody was positive. P-ANCAs were positive to 1/320 by indirect immunofluorescence. Antimyeloperoxidase (anti-MPO) antibodies by enzimoimmunoanalysis were positive to 19U/mL (normal <10) C-ANCA, antiproteinase-3 and anti-glomerular basement membrane antibodies were negative. No monoclonal band was detected in serum. Five days later, an ultrasound-guided renal biopsy was performed with the result of glomerulosclerosis affecting 8 of 11 glomeruli (72%) with a focal segmental necrotizing lesion in one of them. The vessels appeared normal and there was no evidence of vasculitis. A moderate inflammatory infiltrate with polimorfonuclear and lymphocytes was observed in the interstitium. Immunoperoxidase staining was negative for IgA, IgG, IgM, C3, or C4. Red Congo stain for amyloid was negative. A bronchoscopy showed data of pulmonary haemorrhage. A thoracic CT showed some areas with ground-glass appearance. An angiomagnetic nuclear resonance showed a right leg haematoma and thrombosis of the internal saphena. A salivary glands gammagraphy showed a marked asymmetry and decreased uptake of right submandibular gland with normal left glands. Schirmer's test was pathologic (less than 5mm). An echocardiogram showed a light pericardial effusion with hypertrophy of the left ventricle and preserved function. An electromyogram showed data of myopathy. A diagnosis of p-ANCA associated vasculitis was made and treatment with intravenous methylprednisolone (1gm/day) for 3 days was initiated with normalization of renal function and cessation of pulmonary hemorrhage. She refused treatment with cyclophosphamide. During admittance the patient experienced an atrial fibrillation that was resolved with amiodarone and digoxine. The patient was discharged with a creatinine of 1,3mg/dL. Routine visits were performed from this time with normal kidney and pulmonary function testing. Two months later during routine visits the patient complained of the presence of papule-plaques involving the trunk and extremities accompanied by pain in the right hand together with flexion contracture of the 4th and 5th fingers of the right hand. The skin had a brownish appearance, and the arms and the legs were hyperpigmented (figs. 1 y 2). A punch biopsy of the skin showed a preserved epidermis with a fibrosed dermis with numerous collagen bundles and numerous fibroblast-like cells and absence of inflammatory infiltrates or plasma cells. Alcian blue stain demonstrates mucin deposits (fig. 3). A magnetic resonance (MR) of the arm showed an increased signal in the septum of subcutaneous cellular tissue with a “paved” appearance (fig. 4).


.")

At the end of 2004 the patient was readmitted due to a new pulmonary haemorrhage and deteriorated renal function that required corticosteroids and intravenous cyclophosphamide. Skin lesions were unchanged from baseline. She suffered a colonic diverticulitis complicated with perforation and sepsis that ultimately led to death. An autopsy was not carried out.
DiscussionNSF is basically a fibroplasia-an increase in fibrocytes and dermal collagen. Patients present thickened or oedematous skin with indurated papules and plaques involving the extremities and the trunk. The lesions are typically symmetrical. Occasionally swelling of the hands and feet sometimes associated with bulla is noted. Joints contracture may develop very rapidly. Plantar flexion of the feet may be severe enough to make ambulation impossible. Histological findings may vary depending on the stage of the disease development. These findings include thick collagen bundles, variable mucin deposition, and absence of plasma cells and proliferation of dermal fibroblast. CD34-positive dermal dendrocytes are closely apposed to collagen and elastic fibers2. MR imaging shows a heterogeneous pattern of the musculature, as seen in myositis, as well as abnormally inflamed subcutaneous adipose tissue8.
The affected patients were initially identified among recipients of renal transplant, but later cases identified in patients with a variety of different kidney diseases appeared. The severity of NSF is not related to the degree of renal impairment or to the cause of the underlying renal dysfunction. The aetiology and pathogenesis of NSF are unknown, the resemblance to a tissue injury reaction and the presence of myofibroblast, also seen in Spanish toxic olive oil epidemic syndrome, in the tissue specimens suggest that fibrogenic cytokines such as TGF-β may be involved in the evolution of the disease6,13. Decreased level of TGF-β1 after plasmapheresis appears to correlate with the amelioration of this clinical condition4. Hypercoagulability or thrombotic episodes, or both, may precipitate NSF in as many as 12% of the patients10 and anticardiolipin antibodies have been described in some patients6,14. Jimenez et al report a marked elevation of acute phase reactants in 6 patients and a reduced diffusion capacity on pulmonary function testing in 5 patients13. Systemic involvement with fibrosis of diaphragm, psoas muscle or testis3 and increased circulating immune complexes and anti-DNA antibodies have been reported15.
After 2006 numerous published case series have confirmed a very strong associattion of NSF with exposure to Gd-CA. 93% of the uncounfounded biopsy-proven NSF cases linked to a specific Gd-CA have been associated with gadodiamide (Omniscan®). Trasmetillation of the gadolinium chelate because of prolonged clearance of Gd-CA has been proposed as trigger for NSF16 High-dose erythropoietin therapy has been postulated to contribute to the pathogenesis of NSF but our patient did not received treatment with eytrhopoietin17.
The ultimate course of NSF has not been defined. Frequently the disease persists and causes varying degrees of disability. Complete resolution is rare. At present, there is no consistently effective therapy for NSF. Therapeutic modalities that have been attempted up to now include oral and topical steroids, intralesional triamcinolone or methotrexate, cyclosporine, tacrolimus, thalidomide, interferon, photopheresis or plasmapheresis4,10,18. Intravenous immunoglobulin as used in scleromyxoedema have reported slight improvement in joint motion range19.
Differential diagnosis includes scleromyxoedema, which the condition resembles in some aspect, but systemic findings are absent. Scleromyxoedema commonly affects the face, an unusual finding in NSF20. We have not found monoclonal serum paraproteinemia or inflammatory plasma cell infiltrate in the skin biopsy in our patient. Well-circumscribed, indurate, inflamed, hyperpigmented plaques of the lower part of the legs characterize lipodermatosclerosis, usually affecting women who have venous insufficiency and stasis. These features are quiet different from those of our patient. Other disorders to be considered are systemic or localized sclerodermia (morphea), eosinophilic fascitis or the eosinophilia-myalgia syndrome. Our patient developed the characteristic symmetric lesions involving the trunk and extremities without face or neck involvement. There was no Raynaud's phenomenon, sclerodactyly, telangiectasias or perioral furrows as in systemic sclerosis. Morphea is characterized by ivory-colored plaques with violaceous borders and histologically there is increased collagen with the absence of mucin. There was no peripheral–blood eosinophilia, thickening of the deep fascia or infiltrates in the biopsy as seen in eosinophilic fascitis or eosinophilia myalgia syndrome. There was no evidence of amyloid deposition on skin or kidney biopsies. Initially the patient complained of polyarthralgias but arthritis as described in fibroblastic rheumatism was absent. The patient had previously undertaken homeopathic treatment but she did not know the characteristics of the products she had taken. There was no evidence of exposure to toxic oil.
Our patient suffered ARF in the setting of a systemic disease characterized by pulmonary haemorrhage and renal insufficiency together with positive ANA, Ro and p-ANCA antibodies. She suffered a deep venous thrombosis and was treated with subcutaneous heparin and had antecedents of phlebitis but she did not show positive lupus anticoagulant, antiphospholipid antibodies or any abnormal coagulation test. A MR with Gd-Ca (Gadodiamide-Omniscan®) was performed 20 days after admision. The cutaneous lesions began two months later. ANCA associated vasculitis was the final diagnosis in view of the pulmonary haemorrhage and positive anti MPO antibodies, although the findings of the kidney biopsy with marked sclerotic findings were not those typically encountered in Wegener′s granulomatosis or microscopic polyarteritis. The increased prevalence of autoimmune diseases in their family resulted surprising. She had four live daughters, two of them with autoimmune diseases, and other three male pregnancies derived in death in early childhood. No information about the death causes was obtained.
We stress this peculiar case of NSF in a patient with systemic autoimmune disease in an autoimmune family background and the presence of three of the antecedents typically found in NSF: acute renal failure, deep venous thrombosis and exposure to Gd-Ca.


