


Describir las características clínicas, mortalidad y causas de muerte de una serie de pacientes diagnosticados de miositis inflamatoria idiopática del registro REMICAM de la Sociedad de Reumatología de la Comunidad de Madrid (SORCOM).
MétodosEstudio descriptivo retrospectivo multicéntrico de una cohorte de pacientes con diagnóstico de miositis inflamatoria idiopática en seguimiento en servicios de reumatología de hospitales de la Comunidad de Madrid entre enero de 1980 y diciembre de 2014. Se han recogido hasta un total de 313 variables acerca de aspectos demográficos, clínicos y de morbimortalidad, y se ha realizado una comparación entre subgrupos clínicos.
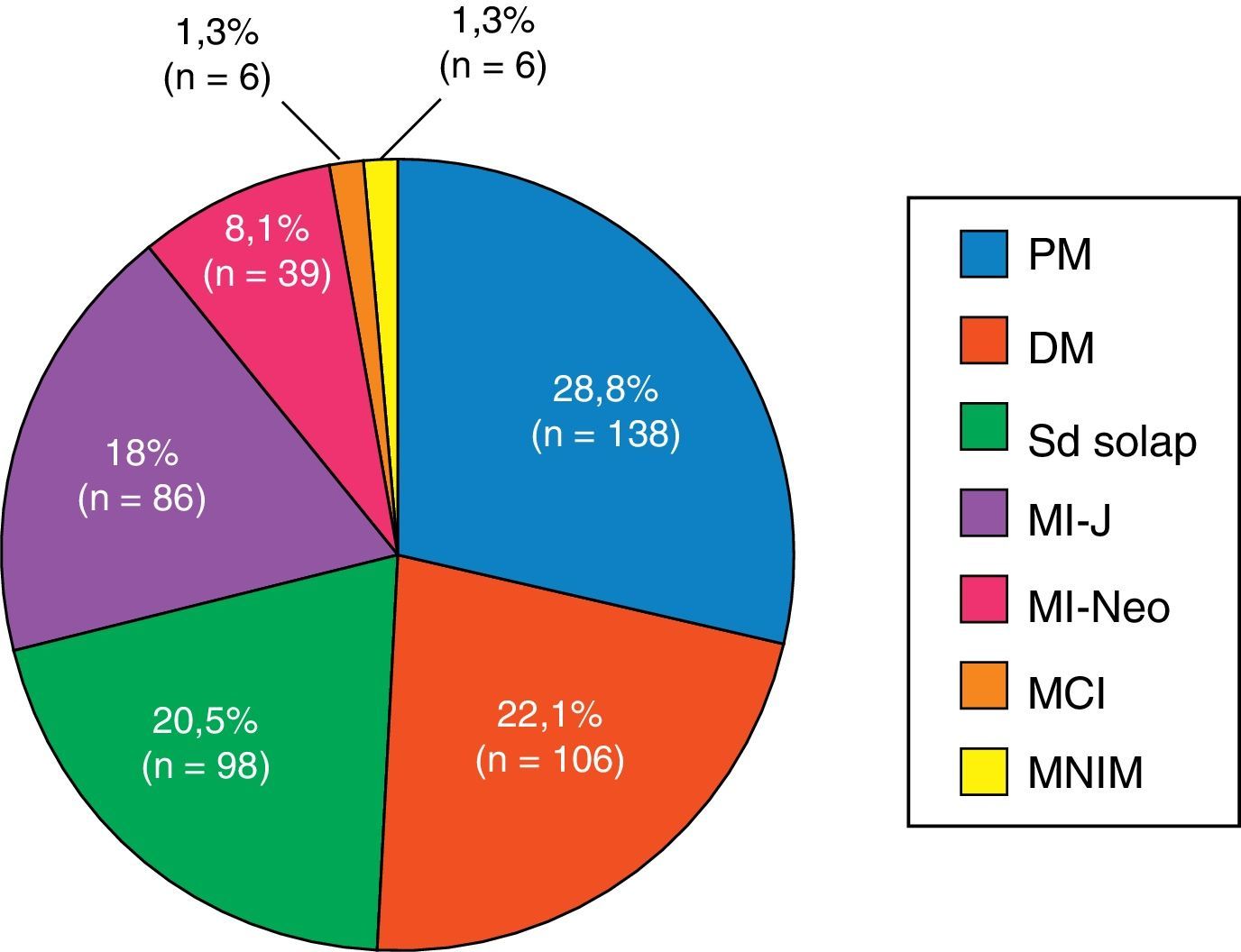
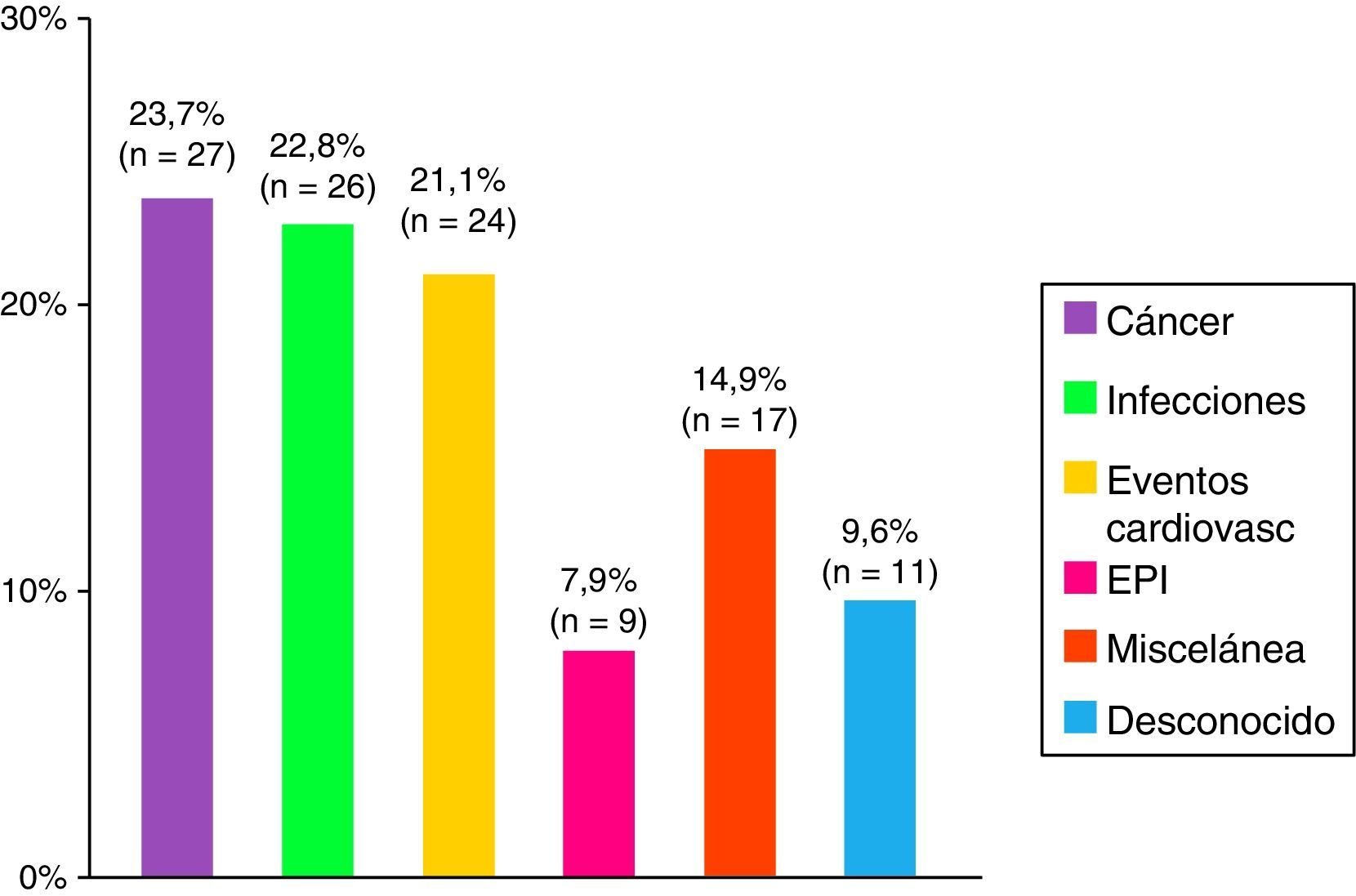
ResultadosSe han reclutado 479 pacientes procedentes de 12 centros, con un 14% de pérdidas durante el periodo de seguimiento. El 74% de los casos eran mujeres, una edad al diagnóstico de 44±23 años, y una media de seguimiento de 10±8 años. Los subgrupos clínicos más frecuentes fueron las formas primarias (PM 29%, DM 22%), seguidas de síndrome de solapamiento (20,5%), miopatías juveniles (18%), miopatías asociadas a cáncer (8%), miopatías necrosantes inmunomediadas (1%) y miositis por cuerpos de inclusión (1%). Durante el periodo de seguimiento se produjeron un total de 114 fallecimientos (28%), siendo las principales causas el cáncer (24%), las infecciones (23%) y los eventos cardiovasculares (21%).
ConclusionesEn el registro REMICAM de miopatías inflamatorias de la Comunidad de Madrid se han reclutado 479 casos de miositis inflamatoria idiopática con datos sociodemográficos, clínicos y pronósticos, suponiendo el mayor registro multicéntrico español en el ámbito de la Reumatología hasta la fecha, y constituyendo una fuente importante para la realización de posteriores subestudios.
To analyze clinical characteristics, survival and causes of death of patients diagnosed with autoimmune inflammatory myositis in the REMICAM registry from the Society of Rheumatology in the Community of Madrid (SORCOM).
MethodsMulticenter cohort of patients diagnosed with autoimmune inflammatory myopathy with follow-up between January 1980 and December 2014. A total of 313 variables concerning demographic, clinical and morbidity data were collected, and a comparison was performed between clinical subgroups.
ResultsA total of 479 patients were recruited from 12 centers, with 14% of patients lost to follow-up. Seventy-four percent of cases were women, age at diagnosis of 44±23 years and a mean follow-up period of 10±8 years. The most frequent clinical subgroups were primary myositis (PM 29%, DM 22%), followed by overlap myositis (20.5%), juvenile myositis (18%), myositis associated with cancer (8%), immune-mediated necrotizing myositis (1%) and inclusion body myositis (1%). During the follow-up period, a total of 114 deaths (28%) were registered, the main causes being cancer (24%), infections (23%) and cardiovascular events (21%).
ConclusionsA total of 479 patients were recruited in the REMICAM registry of inflammatory myopathies. Including sociodemographic, clinical and prognostic information, it represents the largest Spanish multicenter registry to date in rheumatology, and constitutes an important source for conducting further substudies.
Las miopatías inflamatorias idiopáticas (MII) comprenden un grupo heterogéneo de enfermedades sistémicas, caracterizadas por una inflamación no supurativa de la musculatura esquelética y debilidad muscular progresiva, ocasionalmente acompañada de manifestaciones sistémicas. Se considera una enfermedad rara, con una incidencia media global entre uno y 19 casos/millón de habitantes/año1, variando en función de la zona geográfica, los métodos de investigación utilizados y los criterios de clasificación aplicados. Antes del uso generalizado de esteroides y tratamientos inmunosupresores, las tasas de mortalidad eran elevadas2,3, con una mejoría significativa en la supervivencia tras el uso generalizado de estos tratamientos y gracias a un diagnóstico precoz4–6. Las manifestaciones clínicas de las MII, así como su evolución y pronóstico son enormemente heterogéneos, lo que, unido a su baja prevalencia, dificulta el estudio de la enfermedad en ausencia de estudios multicéntricos.
Hasta la fecha, ni en la Comunidad de Madrid ni en España se han realizado estudios multicéntricos de MII en el ámbito de la Reumatología. Por estos motivos, y con el apoyo de la Sociedad de Reumatología de la Comunidad de Madrid (SORCOM), se planteó la formación de un grupo para el Registro de Miopatías inflamatorias en la Comunidad Autónoma de Madrid (REMICAM), para el registro transversal de pacientes, cuya inclusión ya ha finalizado. La disponibilidad de un registro observacional multicéntrico de pacientes con MII podría permitir conocer la realidad de esta enfermedad en la Comunidad de Madrid, así como averiguar la morbimortalidad, o la comparación entre subgrupos de pacientes, en una enfermedad cuya rareza limita la obtención de datos significativos.
Pacientes y métodosObjetivos del estudioLos objetivos principales del REMICAM son describir y caracterizar a los pacientes con MII controlados habitualmente en los servicios de reumatología de la Comunidad de Madrid, con el fin de:
- 1.
Describir las características sociodemográficas y clínicas
- 2.
Estudiar la frecuencia puntual y acumulada de comorbilidad, y las posibles diferencias entre distintos tipos de miopatías
- 3.
Calcular la frecuencia de mortalidad en la serie general y por subgrupos clínicos
Como objetivos secundarios el registro se plantea como un estudio colaborativo multicéntrico para el estudio posterior de aspectos específicos de las miopatías inflamatorias autoinmunes.
Diseño del estudioRegistro multicéntrico retrospectivo de pacientes con MII procedentes de unidades o servicios de reumatología de la Comunidad de Madrid, a partir de un registro transversal, de base hospitalaria, con recogida de la información acerca de datos clínicos, mortalidad y causas de muerte de forma retrospectiva.
Selección de pacientesA través de la SORCOM se remitió invitación para participación en el presente estudio a los distintos servicios o unidades de reumatología de hospitales públicos de la Comunidad de Madrid, así como al servicio de Reumatología del Hospital Madrid Norte de Sanchinarro. Se incluyeron pacientes consecutivos no seleccionados con diagnóstico de MII (dermatomiositis, polimiositis, miopatía por cuerpos de inclusión, miopatía necrosante inmunomediada), en seguimiento en reumatología en algún momento en el periodo comprendido entre enero de 1980 y diciembre de 2014, sin tener en cuenta la edad de inicio del proceso. Los pacientes seleccionados cumplían criterios de Bohan y Peter7,8 y/o criterios de Tanimoto9, y se excluyeron las miopatías de causa tóxica o infecciosa o secundaria a enfermedad neuromuscular. Los pacientes se clasificaron en 7 subgrupos: polimiositis idiopática (PM), dermatomiositis idiopática (DM), miopatías juveniles (MI-J), MII asociada a otra conectivopatía (síndrome de solapamiento), MII asociada a neoplasia maligna (MI-Neo), miositis por cuerpos de inclusión10 y miopatías necrosantes inmunomediadas11. Los pacientes con síndrome de solapamiento debían cumplir criterios de MII y criterios de las siguientes conectivopatías: artritis reumatoide12, esclerosis sistémica13, lupus eritematoso sistémico14, enfermedad mixta del tejido conectivo15o síndrome de Sjögren16. Definimos la miopatía como asociada a neoplasia maligna como aquellos casos en los que el diagnóstico del cáncer se hizo en los 3 años previos o posteriores de la aparición de la miopatía, similar a estudios previos17. Para el estudio comparativo entre los diferentes subgrupos solo se tuvieron en cuenta los 5 primeros subgrupos, dada la baja prevalencia de las miopatías por cuerpos de inclusión y de las miopatías necrosantes inmunomediadas.
VariablesSe incluyeron 313 variables por cada paciente, agrupadas en variables sociodemográficas, de clasificación, mortalidad, comorbilidad, manifestaciones clínicas y analíticas, y datos sobre tratamientos. Todos estos datos se extrajeron de forma retrospectiva de las historias clínicas de los pacientes. Se realizó una exhaustiva monitorización de inconsistencias en los datos de las variables por parte de una empresa externa.
En los datos sociodemográficos se incluyó información sobre sexo, raza, edad al diagnóstico, edad en la última visita (edad del paciente en la última visita disponible del paciente o edad en el momento del fallecimiento del paciente), y tiempo de evolución (tiempo entre fecha del diagnóstico y fecha de la última visita).
Los síntomas generales incluyeron pérdida ponderal>10% o fiebre no explicada por otras causas diferentes a la enfermedad reumatológica. Las manifestaciones cutáneas incluyeron pápulas de Gottron, eritema en heliotropo, signo de Gottron, vasculitis cutánea (definida por biopsia compatible), fotosensibilidad, manos de mecánico, prurito inespecífico, úlcera cutánea, úlcera isquémica en pulpejos de los dedos, eritema periungueal y/o dilatación macroscópica de capilares periungueales, edema en manos y esclerodactilia. Entre las manifestaciones hematológicas comprendieron pacientes con anemia, leucopenia o trombocitopenia atribuidas a la enfermedad, tras exclusión de causas farmacológicas, infecciosas y otras causas. Se tuvieron en cuenta las siguientes manifestaciones digestivas: disfagia, reflujo gastroesofágico (definido como clínica sugestiva y endoscopia digestiva alta y/o pH-metría y/o manometría esofágica demostrando evidencia de dismotilidad del tracto gastrointestinal, tras exclusión de otras causas), hemorragia digestiva alta/baja atribuidas a la enfermedad (definida por hematemesis, melenas o rectorragia, tras exclusión de otras causas) y diarrea o estreñimiento atribuidos a la enfermedad (definidos por clínica compatible, tras exclusión de otras causas). En la enfermedad cardiovascular se incluyeron: enfermedad arterial o tromboembólica venosa o pulmonar, cardiopatía isquémica, arritmias, accidente cerebrovascular o hipertensión pulmonar. La hipertensión pulmonar se definió por ecocardiograma (PSAP estimada≥40mmHg) y/o cateterismo cardíaco (PSAP medida≥25mmHg). La enfermedad pulmonar intersticial se diagnosticó por clínica compatible (disnea de esfuerzo de reciente aparición y/o tos seca y/o fiebre sin otra causa) e imágenes diagnósticas en radiografía de tórax, tomografía axial computarizada pulmonar de alta resolución y/o biopsia pulmonar compatibles, y/o pruebas de función respiratoria compatibles, una vez excluyendo otras causas. La enfermedad pulmonar intersticial se clasificó, cuando fue posible, en función del resultado de la biopsia pulmonar y/o la imagen radiológica, y en caso de discordancia, se llegó a un consenso entre el radiólogo y el anatomopatólogo, en los siguientes subtipos: neumonía intersticial no específica, neumonía intersticial usual, bronquiolitis obliterante con neumonía organizativa, daño alveolar difuso y neumonía criptogénica. Se consideró una infección como grave si precisó hospitalización o si produjo la muerte.
La causa de muerte de los pacientes en seguimiento habitual en su hospital de referencia se tomó según datos de la historia clínica o del certificado de defunción. En caso de pérdida en el seguimiento del paciente, se intentó contactar telefónicamente para conocer el estado del paciente, así como la causa y la fecha de fallecimiento. Las causas de fallecimiento se agruparon en 5 subgrupos en función de la causa subyacente de la muerte: infecciones, evento cardiovascular (arritmia, cardiopatía isquémica, accidente cerebrovascular agudo de origen isquémico hemorrágico, hipertensión pulmonar, insuficiencia cardíaca y tromboembolismo pulmonar), cáncer, enfermedad pulmonar intersticial, y miscelánea.
Análisis estadísticoPara todos los análisis estadísticos se ha prefijado un nivel de significación α de 0,05. Se realizó un análisis estadístico descriptivo básico de factores sociodemográficos, subgrupos de clasificación, comorbilidad y supervivencia. Para la descripción de la muestra se utilizaron medidas de tendencia central (media y mediana) y de dispersión (desviación estándar y rango intercuartílico), así como tablas de frecuencias y distribución de porcentajes para las variables cuantitativas y cualitativas, respectivamente. La comparación entre los diversos tipos de miopatías se llevó a cabo mediante pruebas de diferencia de medias, con análisis de la varianza, y diferencias de proporciones con chi cuadrado y test exacto de Fisher en casos con tamaños de celda inferiores a 5. Todos los análisis se efectuaron con Stata 12.
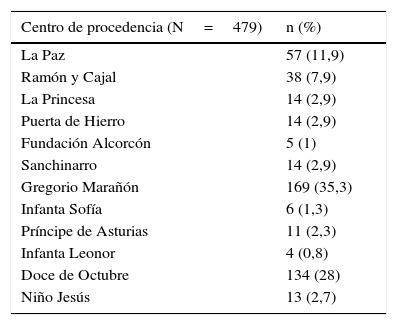
ResultadosLa muestra está formada por 479 pacientes pertenecientes a servicios o unidades de reumatología de 12 hospitales de la Comunidad de Madrid (tabla 1). Setenta pacientes (14,6%) se perdieron en el seguimiento en el momento de inclusión del registro.
Distribución de pacientes por hospitales
| Centro de procedencia (N=479) | n (%) |
|---|---|
| La Paz | 57 (11,9) |
| Ramón y Cajal | 38 (7,9) |
| La Princesa | 14 (2,9) |
| Puerta de Hierro | 14 (2,9) |
| Fundación Alcorcón | 5 (1) |
| Sanchinarro | 14 (2,9) |
| Gregorio Marañón | 169 (35,3) |
| Infanta Sofía | 6 (1,3) |
| Príncipe de Asturias | 11 (2,3) |
| Infanta Leonor | 4 (0,8) |
| Doce de Octubre | 134 (28) |
| Niño Jesús | 13 (2,7) |
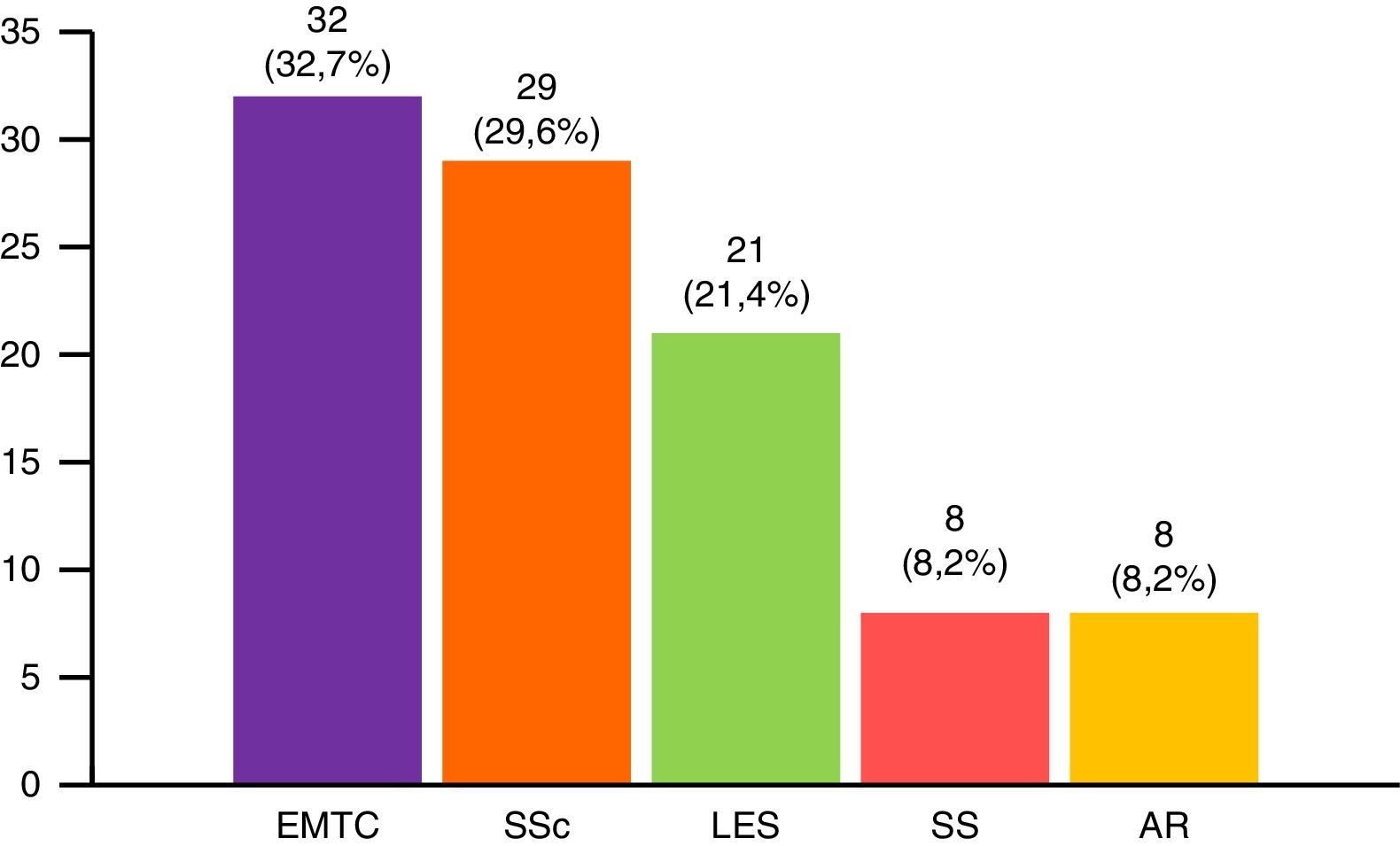
Los subgrupos de miopatías más frecuentes fueron las formas primarias (PM y DM), seguidas del síndrome de solapamiento, miopatías juveniles y miopatías asociadas a neoplasia maligna. Además, existen 6 casos de miopatía por cuerpos de inclusión y 6 pacientes con miopatía necrosante inmunomediada (fig. 1). De los 6 pacientes con miopatía necrosante inmunomediada, solo un caso había estado en tratamiento con estatinas y 3 no. No se pudo obtener esta información de los otros 2 pacientes. La conectivopatía más frecuente asociada a MII en el síndrome de solapamiento fue la enfermedad mixta del tejido conectivo, seguida por la esclerosis sistémica, lupus eritematoso sistémico, síndrome de Sjögren y artritis reumatoide (fig. 2). El 99,6% cumplían criterios de Bohan y Peter, el 97,3% criterios de Tanimoto, y el 96,9% ambos. Según los criterios de clasificación de Bohan y Peter, el 99,6% de los pacientes tendrían un diagnóstico posible, el 94,1% una enfermedad probable y el 67,4% una enfermedad definida. Todos los pacientes con diagnóstico de síndrome de solapamiento excepto un caso presentaron biopsia muscular compatible con miositis o EMG con patrón miopático típico. Dicho paciente se diagnosticó de esclerosis sistémica y presentó debilidad muscular y elevación en niveles de CK al inicio de 748, que mejoró posteriormente con el tratamiento.


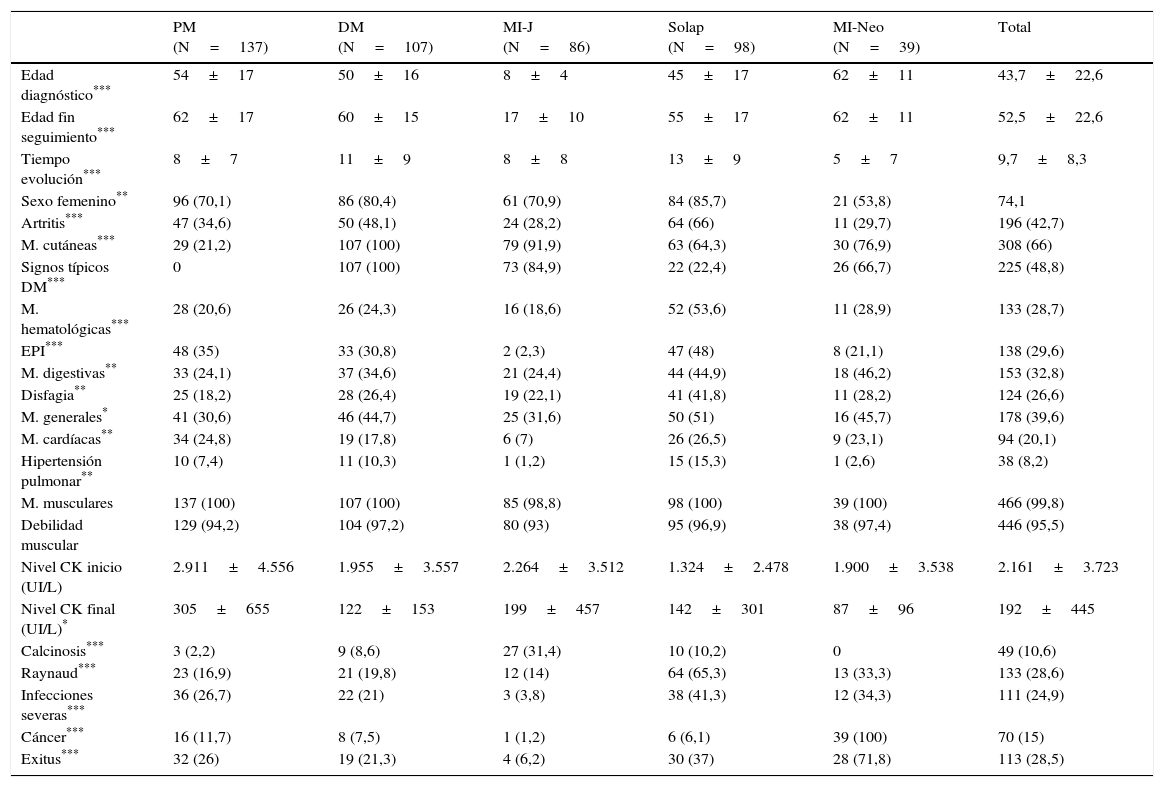
La mayor parte de los casos de la serie son mujeres (74,1%) y de raza caucásica (93,5%). El resto de los datos sociodemográficos vienen reflejados en la tabla 2. En cuanto a las manifestaciones clínicas destaca la elevada prevalencia de enfermedad pulmonar intersticial (29,9%), principalmente de tipo neumonía intersticial usual (28,2%) y neumonía intersticial no específica (23,4%), seguida de bronquiolitis obliterante con neumonía organizativa (4%), daño alveolar difuso (2,4%) y neumonía criptogénica (0,8%), no siendo posible identificar el subtipo en el resto de los pacientes. El 11,6% de los pacientes presentaban niveles de CK por encima de la normalidad al final del seguimiento, destacando la presencia de un tiempo de seguimiento más largo en pacientes con niveles de CK normales (10,3±8,5 años vs. 5,9±5,2 años; p<0,001).
Características clínicas diferenciales según subgrupos clínicos, n (%)
| PM (N=137) | DM (N=107) | MI-J (N=86) | Solap (N=98) | MI-Neo (N=39) | Total | |
|---|---|---|---|---|---|---|
| Edad diagnóstico*** | 54±17 | 50±16 | 8±4 | 45±17 | 62±11 | 43,7±22,6 |
| Edad fin seguimiento*** | 62±17 | 60±15 | 17±10 | 55±17 | 62±11 | 52,5±22,6 |
| Tiempo evolución*** | 8±7 | 11±9 | 8±8 | 13±9 | 5±7 | 9,7±8,3 |
| Sexo femenino** | 96 (70,1) | 86 (80,4) | 61 (70,9) | 84 (85,7) | 21 (53,8) | 74,1 |
| Artritis*** | 47 (34,6) | 50 (48,1) | 24 (28,2) | 64 (66) | 11 (29,7) | 196 (42,7) |
| M. cutáneas*** | 29 (21,2) | 107 (100) | 79 (91,9) | 63 (64,3) | 30 (76,9) | 308 (66) |
| Signos típicos DM*** | 0 | 107 (100) | 73 (84,9) | 22 (22,4) | 26 (66,7) | 225 (48,8) |
| M. hematológicas*** | 28 (20,6) | 26 (24,3) | 16 (18,6) | 52 (53,6) | 11 (28,9) | 133 (28,7) |
| EPI*** | 48 (35) | 33 (30,8) | 2 (2,3) | 47 (48) | 8 (21,1) | 138 (29,6) |
| M. digestivas** | 33 (24,1) | 37 (34,6) | 21 (24,4) | 44 (44,9) | 18 (46,2) | 153 (32,8) |
| Disfagia** | 25 (18,2) | 28 (26,4) | 19 (22,1) | 41 (41,8) | 11 (28,2) | 124 (26,6) |
| M. generales* | 41 (30,6) | 46 (44,7) | 25 (31,6) | 50 (51) | 16 (45,7) | 178 (39,6) |
| M. cardíacas** | 34 (24,8) | 19 (17,8) | 6 (7) | 26 (26,5) | 9 (23,1) | 94 (20,1) |
| Hipertensión pulmonar** | 10 (7,4) | 11 (10,3) | 1 (1,2) | 15 (15,3) | 1 (2,6) | 38 (8,2) |
| M. musculares | 137 (100) | 107 (100) | 85 (98,8) | 98 (100) | 39 (100) | 466 (99,8) |
| Debilidad muscular | 129 (94,2) | 104 (97,2) | 80 (93) | 95 (96,9) | 38 (97,4) | 446 (95,5) |
| Nivel CK inicio (UI/L) | 2.911±4.556 | 1.955±3.557 | 2.264±3.512 | 1.324±2.478 | 1.900±3.538 | 2.161±3.723 |
| Nivel CK final (UI/L)* | 305±655 | 122±153 | 199±457 | 142±301 | 87±96 | 192±445 |
| Calcinosis*** | 3 (2,2) | 9 (8,6) | 27 (31,4) | 10 (10,2) | 0 | 49 (10,6) |
| Raynaud*** | 23 (16,9) | 21 (19,8) | 12 (14) | 64 (65,3) | 13 (33,3) | 133 (28,6) |
| Infecciones severas*** | 36 (26,7) | 22 (21) | 3 (3,8) | 38 (41,3) | 12 (34,3) | 111 (24,9) |
| Cáncer*** | 16 (11,7) | 8 (7,5) | 1 (1,2) | 6 (6,1) | 39 (100) | 70 (15) |
| Exitus*** | 32 (26) | 19 (21,3) | 4 (6,2) | 30 (37) | 28 (71,8) | 113 (28,5) |
DM: dermatomiositis idiopática; EPI: enfermedad pulmonar intersticial; MI-J: miopatía juvenil; MI-Neo: miopatía asociada a neoplasia maligna; PM: polimiositis idiopática; Solap: síndrome de solapamiento.
Las edades y tiempo de evolución y niveles de CK están expresadas como media±DE; el resto de variables vienen expresados como número y porcentaje (en paréntesis).
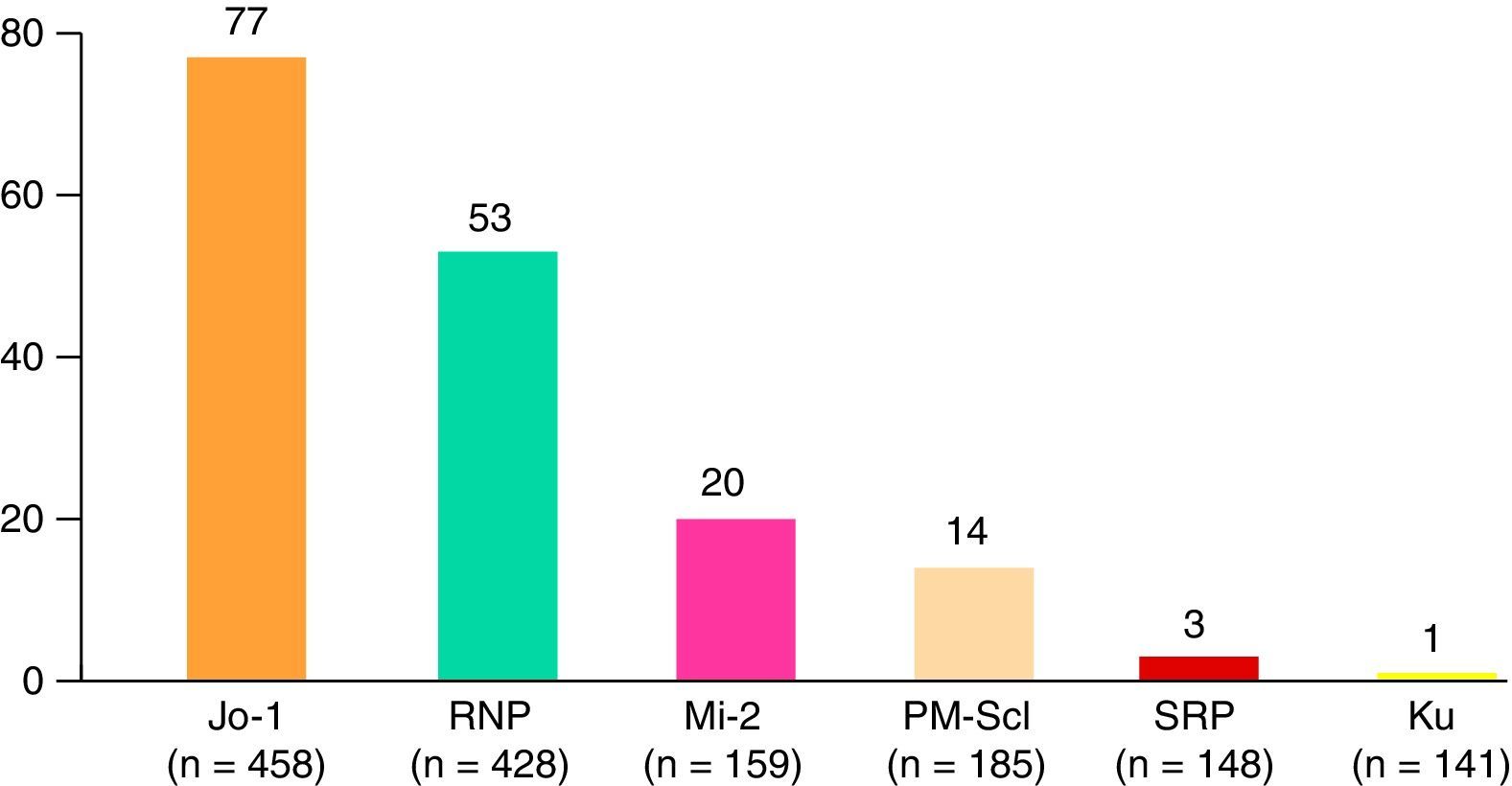
Los pacientes con DM tuvieron más síntomas generales en comparación con las PM, más manos de mecánico y más manifestaciones cutáneas típicas. Las miopatías juveniles se caracterizaron por presentar más manifestaciones cutáneas típicas de las dermatomiositis, pero menos fenómeno de Raynaud, manifestaciones extramusculares, neoplasias malignas e infecciones graves. Por otra parte, en el síndrome de solapamiento fueron más comunes la artritis, el fenómeno de Raynaud, las manifestaciones hematológicas, la enfermedad pulmonar intersticial, las manifestaciones sistémicas, neuropsiquiátricas, y renales. Las miopatías paraneoplásicas presentaron un tiempo más corto de seguimiento y unos niveles más bajos de CK al final del seguimiento comparado con el resto de los subgrupos. Los anticuerpos específicos de miositis más frecuentes fueron los anti-Jo1 (77 casos), seguido de anticuerpos anti-RNP, anti-Mi2, anti-PM-Scl y otros menos frecuentes (fig. 3). El resto de las manifestaciones clínicas vienen reflejadas en la tabla 2.
Comorbilidad y complicaciones
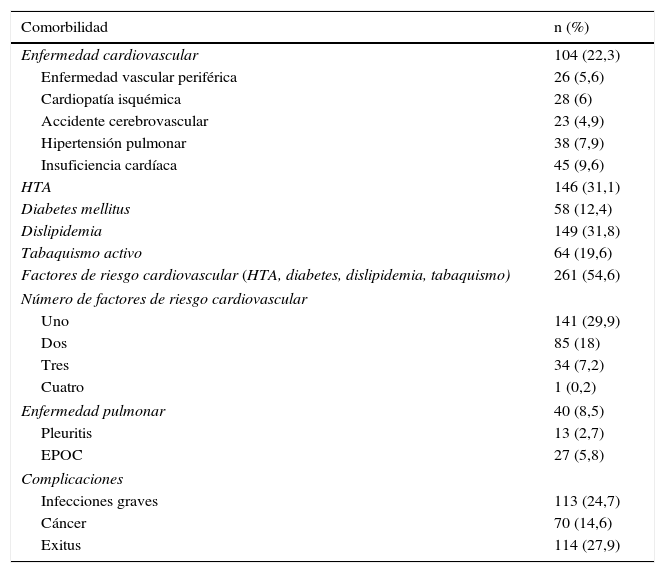
El 54,6% de los pacientes presentaron algún factor de riesgo cardiovascular, principalmente dislipidemia, hipertensión arterial, diabetes mellitus y tabaquismo activo, y el 22,3% se complicaron con algún evento cardiovascular (tabla 3).
Comorbilidad y complicaciones
| Comorbilidad | n (%) |
|---|---|
| Enfermedad cardiovascular | 104 (22,3) |
| Enfermedad vascular periférica | 26 (5,6) |
| Cardiopatía isquémica | 28 (6) |
| Accidente cerebrovascular | 23 (4,9) |
| Hipertensión pulmonar | 38 (7,9) |
| Insuficiencia cardíaca | 45 (9,6) |
| HTA | 146 (31,1) |
| Diabetes mellitus | 58 (12,4) |
| Dislipidemia | 149 (31,8) |
| Tabaquismo activo | 64 (19,6) |
| Factores de riesgo cardiovascular (HTA, diabetes, dislipidemia, tabaquismo) | 261 (54,6) |
| Número de factores de riesgo cardiovascular | |
| Uno | 141 (29,9) |
| Dos | 85 (18) |
| Tres | 34 (7,2) |
| Cuatro | 1 (0,2) |
| Enfermedad pulmonar | 40 (8,5) |
| Pleuritis | 13 (2,7) |
| EPOC | 27 (5,8) |
| Complicaciones | |
| Infecciones graves | 113 (24,7) |
| Cáncer | 70 (14,6) |
| Exitus | 114 (27,9) |
EPOC: enfermedad pulmonar obstructiva crónica; HTA: hipertensión arterial.
Un 25% de los pacientes presentaron complicaciones en forma de infecciones graves, principalmente infecciones respiratorias bajas (63%), seguidas de urinarias (14%), gastrointestinales (10%) y otras causas menos frecuentes. En un 30% de los casos de infecciones graves el desenlace fue la muerte. Se complicaron más frecuentemente con infecciones graves aquellos pacientes con síndrome de solapamiento y miopatías asociadas a neoplasia maligna. En cuanto a las miopatías asociadas a cáncer, los tumores más frecuentes fueron cáncer de pulmón y cutáneo (8 cada uno), linfoma (7) y mama (6). En 2 casos no se llegó a saber el origen del cáncer.
Exitus y causas de muerteDurante el periodo de estudio se produjo un total de 114 fallecimientos, lo que supone el 28% de la muestra de análisis, siendo las principales causas el cáncer, las infecciones y los eventos cardiovasculares (fig. 4). Los pacientes con miopatías asociadas a cáncer presentaron un peor pronóstico, seguido del síndrome de solapamiento, mientras que los pacientes con miopatías juveniles presentaron un mejor pronóstico.
Tratamientos de fondo
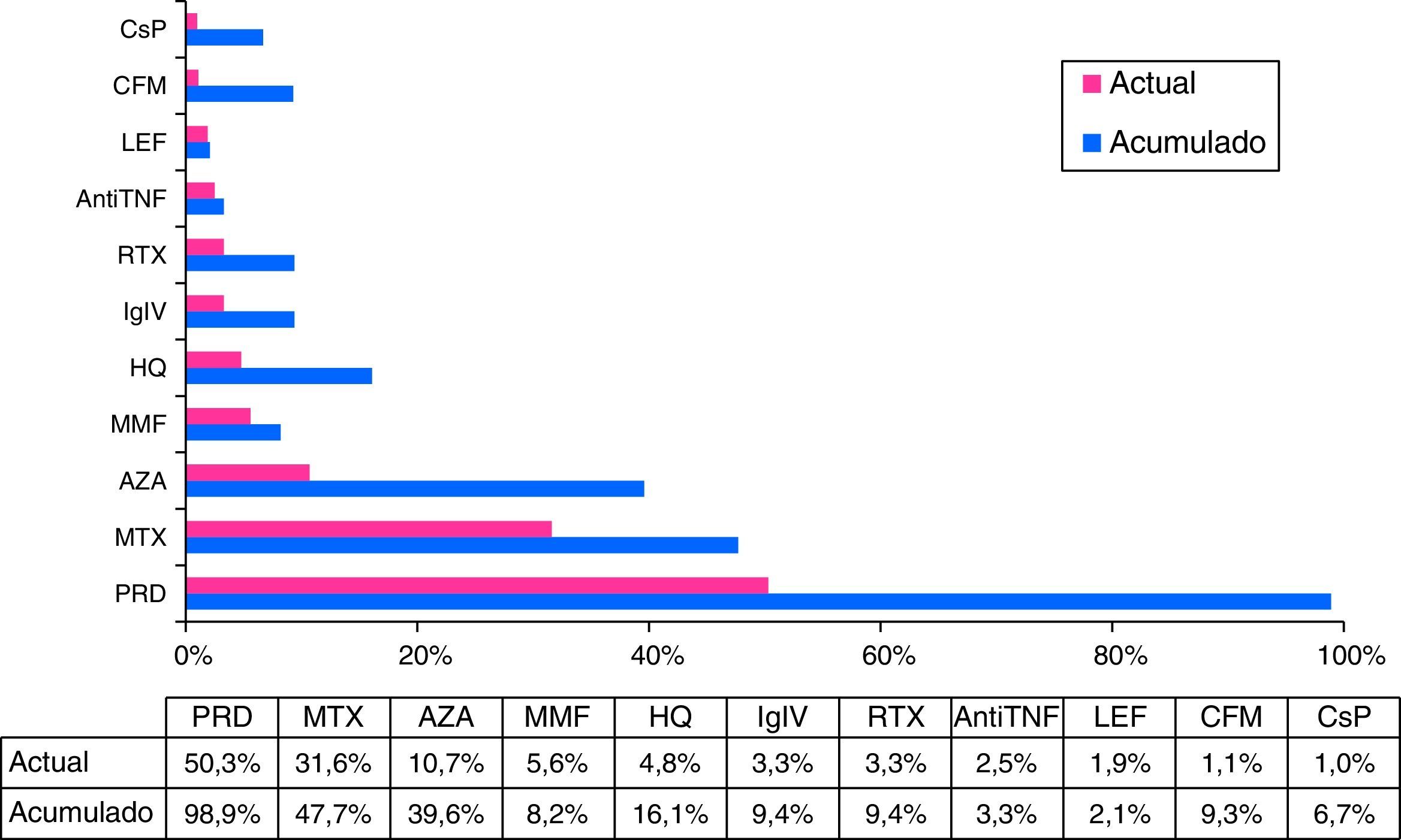
En relación con los tratamientos de fondo, una parte muy importante de los pacientes recibieron glucocorticoides intravenosos (15%) u orales (99%) en alguna ocasión, y la mitad de ellos (50,3%) seguían precisando esteroides orales en la última visita efectuada, con una dosis menor de 30mg/día en el 95% de los casos al final del seguimiento. El 13,2% de los pacientes precisaron más de un ciclo de esteroides orales durante el tiempo de evolución de la enfermedad. Se utilizaron inmunosupresores en el 74,1% de los casos, de los cuales el 30,5% precisaron el uso de 2 o más fármacos durante la evolución de la enfermedad (con una media de 2,4±0,9 fármacos inmunosupresores en estos casos). Los tratamientos de fondo no biológicos más utilizados, en porcentaje acumulado, fueron el metotrexato (48%), azatioprina (40%), hidroxicloroquina (16%) y ciclofosfamida (9%). Un 9% fueron tratados con inmunoglobulina intravenosa y un 12% recibieron tratamiento biológico en alguna ocasión (siendo el más frecuente el rituximab en un 9% de los casos) (fig. 5).

Tratamientos de fondo acumulados y actuales.
AntiTNF: tratamiento anti-TNFα; AZA: azatioprina; CFM: ciclofosfamida; CsP: ciclosporina A; HQ: hidroxicloroquina; IgIV: inmunoglobulinas intravenosas; LEF: leflunomida; MMF: micofenolato mofetilo; MTX: metotrexato; PRD: prednisona; RTX: rituximab.
Este es el primer registro multicéntrico de miopatías inflamatorias en la Comunidad de Madrid, y el que presenta la recopilación de datos más extensa hasta el momento. El registro se realizó con el objetivo de analizar de forma retrospectiva las características clínicas, comorbilidad y complicaciones de las miopatías inflamatorias de origen autoinmune en un estudio multicéntrico de 12 hospitales de la Comunidad de Madrid. Se realizó una comparación de las características clínicas, analíticas y el pronóstico entre los diferentes subgrupos de miopatías inflamatorias de origen autoinmune.
Los criterios de Tanimoto9, publicados en 1995, aportan una mayor especificidad a los de Bohan y Peter18,19. Los únicos 2 casos que no cumplían criterios de Bohan y Peter pero sí de Tanimoto corresponden a 2 pacientes con síndrome por anticuerpos de antisintetasa, cursando con elevación de enzimas musculares, artritis, síntomas generales, y enfermedad pulmonar intersticial en ambos casos, y manos de mecánico en uno de los casos.
Las MII pueden aparecer asociadas a diversas enfermedades del tejido conectivo, describiéndose con una frecuencia muy variable, entre el 7% y el 60% según las series6,20–23. En el presente estudio se emplearon los criterios de clasificación habitualmente aceptados para concluir que realmente los pacientes tenían una conectivopatía asociada a miopatía inflamatoria12–16. Un 21% de los casos se clasificaron como síndrome de solapamiento, siendo esta prevalencia muy similar a la de una serie húngara20, aunque superior a otra serie española procedente de un centro hospitalario6. Parte de los pacientes de esta serie fueron incluidos en el presente registro. Las diferencias observadas en la mayor proporción de pacientes con síndrome de solapamiento en el REMICAM respecto de la otra serie española pudieran deberse al desarrollo de nuevas manifestaciones en esos pacientes que hayan permitido diagnosticarlos de otras conectivopatías asociadas, como ha sido propuesto por otros autores24, o a diferencias en las características de los demás pacientes incluidos.
Únicamente el 11,6% de los pacientes presentaban unos niveles de CK por encima de la normalidad al final del seguimiento, lo cual puede dar una idea algo aproximada de los pacientes en actividad mantenida. No obstante, la recogida de otros datos adicionales, como los propuestos por el grupo International Myositis Outcome Assessment Collaborative Study Group (IMACS) y el Paediatric Rheumatology International Trials Organisation (PRINTO) para la definición de actividad de la enfermedad, tales como la medición de la fuerza muscular, escalas analógicas visuales de la enfermedad global del paciente/padres y médico, cuestionarios sobre función física y otros datos, podrían haber dado una idea más exacta de la actividad de la enfermedad25,26. Sin embargo, el estudio retrospectivo limita la obtención de estos datos, así como los datos para la evaluación de recaída de la enfermedad.
Como era esperable respecto a las diferencias clínicas entre los diferentes subgrupos, los pacientes con síndrome de solapamiento presentaron una enfermedad diferente, con síntomas característicos de otras enfermedades, principalmente esclerosis sistémica o lupus eritematoso sistémico, similar al resultado de otras series6. En cuanto a la comorbilidad asociada destaca que casi la mitad de los pacientes presentaron algún factor de riesgo cardiovascular, siendo el más frecuente la dislipidemia, seguido de la hipertensión arterial.
La incidencia de cáncer en las miositis varía ampliamente entre los diferentes estudios publicados27,28, dependiendo principalmente de la definición del subgrupo de miopatía asociada a cáncer, así como de la edad y sexo de los pacientes, y de las características de las series. La cohorte REMICAM presenta una incidencia más baja respecto a la mayor parte de otras series publicadas. Esto probablemente sea debido a la definición de miopatía asociada a cáncer, así como a la inclusión de pacientes con síndrome de solapamiento y al menor porcentaje de casos de DM, que clásicamente se han relacionado con el mayor riesgo de cáncer6,17.
La mortalidad global en REMICAM fue del 28,5%, similar a otra serie española y asiática, pero más elevada que en otras series6,29. La causa más frecuente de muerte fue el cáncer, seguido de cerca por las infecciones y los eventos cardiovasculares, similar a otras series30,31. En la mayor parte de las series la mortalidad depende de los criterios de inclusión utilizados, de las enfermedades asociadas, del tiempo de seguimiento de los pacientes, y de forma importante, de la inclusión o exclusión de miopatías asociadas a cáncer, que ensombrecen el pronóstico. Así, la mayor supervivencia de algunas series puede deberse a la exclusión de miopatías asociadas a cáncer, a la mayor proporción de casos con formas juveniles, a la menor edad de los pacientes y a la elevada proporción de pérdidas de los estudios4,5.
El estudio es susceptible de presentar sesgos de selección, hecho fundamentalmente relacionado con el método de selección de centros. Es posible, por ejemplo, que los enfermos de los centros interesados en participar fueran más graves o estén mejor estudiados. Sin embargo, este sesgo se ve limitado por el hecho de que el presente estudio estuviera abierto también a centros de especialidades, donde previsiblemente habría casos más benignos, así como por el hecho de que las MII son enfermedades de larga evolución y con una elevada morbilidad, lo cual hace probable que la mayor parte de los casos sean derivados a centros terciarios en algún momento durante la evolución de la enfermedad.
Por otra parte, el diseño retrospectivo del estudio impide el control sobre la calidad de los datos, facilita la aparición de errores de medición y la disponibilidad de información sobre importantes variables de confusión. Sin embargo, la baja proporción de pérdidas de pacientes y la exhaustiva monitorización de inconsistencias por parte de una empresa externa hace poco probable que esta limitación afecte de forma importante a la calidad de los resultados.
Hasta el momento REMICAM es el mayor registro de pacientes con miopatías en la Comunidad de Madrid y a nivel nacional en el ámbito de la Reumatología, con estudio detallado de características clínicas, morbimortalidad y subgrupos clínicos. Actualmente está pendiente la realización de más subestudios que permitan un mayor conocimiento sobre la evolución de esta enfermedad.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónEl presente trabajo ha sido parcialmente financiado por una beca SORCOM-MSD.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la junta directiva de la SORCOM, y en especial a Santos Castañeda y a Ana Cruz, por su apoyo incondicional para poder llevar a cabo este proyecto; a Loreto Carmona por sus consejos y orientación en la metodología; y a la SER, por el apoyo logístico para las reuniones del grupo.


