





To assess the effectiveness and safety of certolizumab pegol (CZP) in Spanish patients with RA.
Materials and methodsSONAR (NCT01526434), a 12-week, open-label, prospective, observational, multicenter study. Patients with active RA for ≥3 months, according to ACR criteria, were treated with CZP (400mg at Weeks 0, 2 and 4, then 200mg every 2 weeks). The primary effectiveness endpoint was change from baseline (CFB) in Health Assessment Questionnaire-Disability Index (HAQ-DI) at Week 12. Other assessments included DAS28(ESR), patient's assessment of arthritis pain (PtAAP-VAS) and Short Form 36-item Health Survey (SF-36) physical component summary (PCS) and mental component summary (MCS). Joint inflammation was investigated using Power Doppler (PD) ultrasound (US), to detect effusion, synovial hypertrophy and synovial PD signal. PDUS outcomes assessed CFB to Week 12 in synovial hypertrophy, effusion and PD signal indices.
ResultsA total of 77/80 enrolled patients received ≥1 dose of CZP. The 12-week mean reduction from baseline (SD) was −0.6 (0.6) for HAQ-DI and −2.2 (1.5) for DAS28(ESR). PtAAP-VAS was reduced from baseline (mean [SD]: −36.8 [26.8]) and improvements in SF-36 PCS and SF-36 MCS were reported. Synovial hypertrophy, effusion and PD signal indices were reduced from baseline to Week 12. One death was reported during the study.
ConclusionsSpanish patients with RA demonstrated improvements in clinical, PDUS and patient-reported outcomes over 12 weeks of CZP treatment. No new safety signals were identified, and the safety profile was in line with previous CZP studies. These results support previous clinical trial findings investigating CZP treatment for active RA.
Evaluar la eficacia y la seguridad de certolizumab pegol (CZP) en pacientes españoles con artritis reumatoide (AR).
Materiales y métodosSONAR (NCT01526434), un estudio multicéntrico, observacional, prospectivo, abierto de 12 semanas. Pacientes con AR activa ≥3 meses, según criterios ACR, recibieron CZP (400mg en las semanas 0, 2 y 4, seguido de 200mg cada 2 semanas). La variable principal de eficacia fue el cambio desde el inicio (CDI) en el HAQ en la semana 12. Otras evaluaciones incluían el DAS28-VSG, la valoración del dolor (PtAAP-VAS) y el componente físico (PCS) y mental (MCS) del SF-36. La inflamación articular se estudió utilizando ecografía con Power Doppler (PDUS) midiendo derrame, hipertrofia sinovial y señal PD sinovial. Los resultados de PDUS evaluaron el CDI hasta la semana 12 en índices de hipertrofia sinovial, derrame y PD.
ResultadosUn total de 77/80 pacientes recibieron ≥una dosis de CZP. La reducción media en 12 semanas desde el inicio (DE) fue de −0,6 (0,6) para HAQ y de −2,2 (1,5) para DAS28-VSG. La PtAAP-VAS disminuyó desde el inicio (media [DE]: −36,8 [26,8]) y hubo mejorías en los componentes PCS y MCS del SF-36. Los índices de señales de hipertrofia sinovial, derrame y PD disminuyeron desde el inicio hasta la semana 12. Se notificó una muerte durante el estudio.
ConclusionesLos pacientes españoles con AR mostraron mejoras en resultados clínicos, PDUS y notificados por el paciente durante 12 semanas de tratamiento con CZP. No hubo nuevas señales de seguridad, y el perfil de seguridad estaba en línea con estudios previos. Estos resultados respaldan los hallazgos de ensayos clínicos previos de CZP en AR.
Rheumatoid arthritis (RA) is a chronic, typically progressive, autoimmune disorder characterized by polyarticular synovial inflammation. It is responsible for functional disability, joint destruction and significant physical pain.1 Symptoms arise as a result of excessive production of pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, by activated T-cells.2
Treatment options for RA include biological agents and, during the period of the described trial, the biologics approved for the treatment of RA in Spain included abatacept, adalimumab, anakinra, certolizumab pegol (CZP), etanercept, golimumab, infliximab, rituximab and tocilizumab.3 Improvements in clinical outcomes have been demonstrated in patients with RA using biologic agents including anti-TNFs.4–6 However, large proportions of patients still discontinue anti-TNF therapies as a result of failure to reach an adequate clinical response (primary failure), loss of clinical response (secondary failure), or as a result of adverse events.7
Patients experiencing a lack of effectiveness or adverse events may be switched to an alternative anti-TNF, however, the clinical practice guideline, available during the described trial, did not confirm whether a second anti-TNF, or a biologic with an alternative mechanistic route, would be favorable, due to insufficient data.8 Establishing the effectiveness of anti-TNFs in patients who have experienced an inadequate response to prior treatment is of importance since reduced response to subsequent biological treatment has been shown to be proportional to increased prior use of biologics.9 This could assist physicians in making the most appropriate next choice of treatment for their patients.
Ultrasound (US) is a noninvasive, low-cost imaging technique which is well accepted by patients, not affected by metallic implants or prostheses, and can be repeated as many times as is necessary.10 US is recommended for the detection of synovitis, effusion and erosions when this information is considered to be clinically relevant for the therapeutic management of patients. This recommendation is according to the clinical practice guideline for rheumatoid arthritis management (GUIPCAR), published in 2016, which was endorsed by the Spanish Society of Rheumatology.11 Power Doppler ultrasound (PDUS) evaluates subclinical synovitis by visualizing synovial inflammatory joint changes that may previously have gone undetected by conventional clinical and radiographic examinations. This can facilitate physicians’ assessment of true inflammation, a typical precursor to the establishment of clinically detectable RA.12
This 12-week, open-label, observational study assessed the impact of treatment with CZP on clinical, patient-reported, and US outcomes in Spanish patients with active RA (with or without prior anti-TNF exposure).
Materials and methodsPatientsPatients were aged ≥18 years old with active RA (determined according to ACR 1987 criteria)13 of over 3 months duration, with 28-joint Disease Activity Score (DAS28) (erythrocyte sedimentation rate [ESR]) >4.5 and C-reactive protein (CRP) >1.0mg/dL at baseline. Additionally, all patients had previously been treated with synthetic disease modifying anti-rheumatic drugs (DMARDs). Patients either had no other prior anti-TNF treatment (naïve patient) or CZP was administered after failure of the first anti-TNF treatment (first-switch patient). Patients enrolled onto the study were actively receiving CZP treatment and were not required to have a wash-out period prior to enrollment. The safety set (SS) consisted of all enrolled patients who took at least 1 dose of CZP. The full analysis set (FAS) consisted of all patients in the SS who had at least 1 valid baseline and post-baseline effectiveness assessment for any effectiveness variable and who had no important protocol deviations. Patients identified as having important protocol deviations were those not initiating CZP treatment, or not fulfilling the study inclusion criteria outlined above.
Study designSONAR (NCT01526434) was a 12-week, open-label, prospective, observational, multicenter, post-marketing study. Both naïve and first switch patients were treated with CZP (400mg at Weeks 0, 2 and 4 followed by 200mg every 2 weeks). CZP was administered subcutaneously, either by patients or according to the standard clinical setting for the prescribing physician and as defined by the Summary of Product Characteristics (SmPC). Permitted concomitant treatments included methotrexate, corticosteroids and analgesics including nonsteroidal anti-inflammatory drugs (NSAIDs). Assessments were carried out at weeks 0, and 12, with a 16-week safety follow-up visit. All patients provided written consent to participate in this study, which was carried out in accordance with local regulations, the Declaration of Helsinki and the local laws of Spain.
Study assessmentsThe primary effectiveness endpoint was the mean change from baseline in Health Assessment Questionnaire-Disability Index (HAQ-DI) for RA,14 at Week 12 in the FAS population; this population was also grouped by prior anti-TNF experience to compare HAQ-DI scores between anti-TNF naïve and first switch patients from baseline to Week 12. Other assessments included DAS28 (ESR),15 Patient's Assessment of Arthritis Pain (PtAAP) – Visual Analog Scale (VAS)16 and Short Form 36-item Health Survey (SF-36) physical component summary (PCS) and mental component summary (MCS).17 For effectiveness assessments, the data were assigned to a visit based on the actual date of the routine clinical visit. Day was calculated relative to the baseline visit (recorded at Week 0). Any visit between Day ≥63 and ≤98 was assigned as Week 12 for the purposes of the analysis.
PDUS assessment involved the detection and grading (from 0 to 3) of gray-scale synovitis and Power Doppler (PD) synovial signal of 24-joints (12-joint US assessment for RA18 and the bilateral 4 and 5 metacarpophalangeal [MCP] joints and 2 to 5 metatarsophalangeal joints). Synovial hypertrophy and effusion were graded, semi-quantitatively, from 0 to 3, where 0=absence; 1=mild; 2=moderate; and 3=marked. PD signal was graded according to the following scale: 0=absence, no synovial flow; 1=mild, ≤3 isolated signals; 2=moderate, >3 isolated signals or confluent signal in less than half of the synovial area; 3=marked, signals in more than half of the synovial area.19 Assessments were made according to the Outcome Measures in Rheumatology definitions and published scoring systems.20 An index for effusion, synovial hypertrophy and synovial PD signal was calculated by summing the corresponding scores from each assessed joint. PDUS outcomes measured included change from baseline to Week 12 in global US 24-joint indices (including synovial hypertrophy index, effusion index, and PD signal index). Intra-reader reliability was monitored by evaluating the first two patients at each center twice (2–4 days between evaluations) before the treatment period and was tested on representative images of joints included in the study. Intra-class correlation coefficients are reported for synovial hypertrophy index, effusion index, and PD signal index and were based on the repeated assessments at baseline for these patients.
Adverse drug reactions (ADRs), were recorded and treatment-emergent adverse drug reactions (TEADRs) were defined as ADRs if they started on or after the date of first study medication and up to 70 days after the last (most recent) dose of CZP. Adverse events were classified as ADRs when a causal relationship between the product and the occurrence was suspected by the reviewing healthcare professional. TEADRs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1.
Statistical analysisThe sample size was calculated based on the precision of the 95% confidence interval (CI) around the expected HAQ-DI score mean change from Baseline to Week 12. A difference of at least ±0.22 was assumed to be sufficient to recognize a minimal clinically important difference (MCID) in HAQ-DI score, and the proportion of patients achieving MCID in HAQ-DI was calculated post hoc.21 A sample size of 95 patients was estimated to provide at least 90% of statistical power to establish a difference of 0.22 as significant with a SD of 0.6 in a two-sided one-sample t-test, with a significance level of 0.05. A HAQ-DI difference greater than 0.22 could be confirmed with fewer patients.
The mean changes in HAQ-DI scores from Baseline to Week 12 were analyzed by comparing the values at Baseline and Week 12 using a paired t-test. The primary analysis was run based on last observation carried forward (LOCF) imputation of missing data from dropouts. A paired t-test was also used to compare Baseline and Week 12 values for SF-36, PtAAP-VAS and DAS28(ESR). Apart from the p-value related to the primary endpoint, all p-values are nominal.
Statistical analysis and generation of tables, figures and patient data listings were performed using Statistical Analysis System (SAS®) Version 9.4. P-values are only provided for the mean change from baseline, to Week 12, in HAQ-DI (the primary endpoint), DAS28(ESR), PtAAP and SF36 PCS and MCS, all other variables are represented by CIs. Nominal p values were not calculated for comparisons between the patients split by prior anti-TNF exposure, due to the small sample number, therefore data are shown as the mean and 95% CI for HAQ-DI and PDUS outcomes for each subgroup.
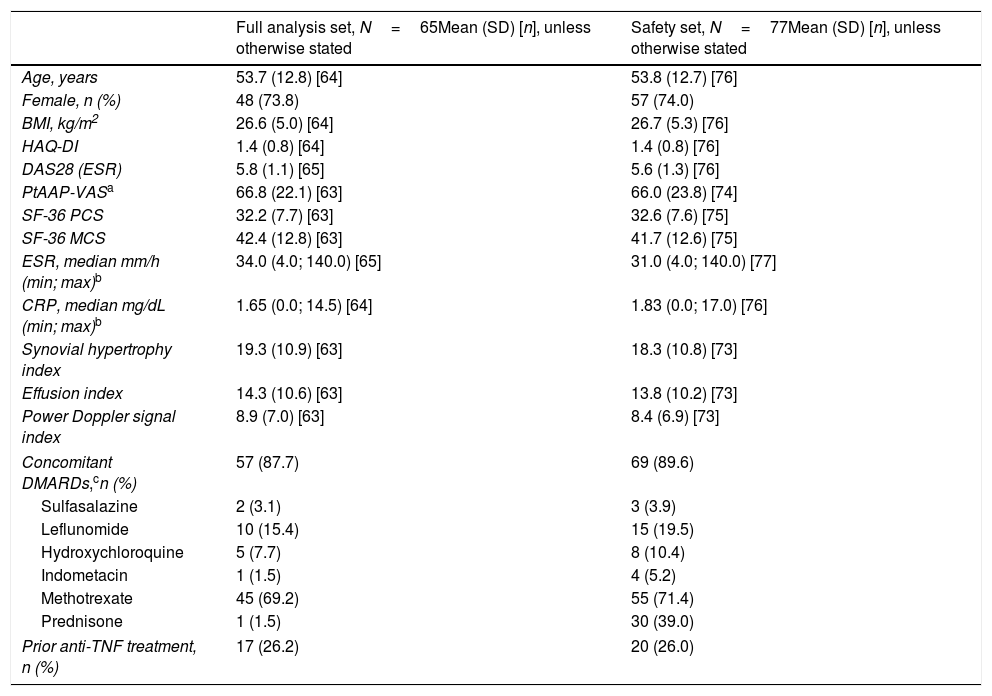
ResultsPatient disposition and baseline characteristicsA total of 77/80 enrolled patients received at least one dose of CZP during the trial and were included in the SS (Table 1), of which 65 patients provided at least one valid baseline and post-baseline effectiveness assessment, without any important protocol deviations during the study (FAS); the remaining 12 recruited patients did not meet these criteria for inclusion within the FAS. The reasons for excluding these patients from the FAS were: 3 patients did not have active RA (defined as DAS28(ESR)>4.5 and CRP>1.0mg/dL) at baseline, prior anti-TNF use was unknown for 1 patient, and 9 patients did not have a valid baseline and post-baseline effectiveness assessment. At baseline, 57/77 (74%) patients within the SS and 48/65 (73.8%) patients in the FAS were anti-TNF naïve. Of patients in the FAS, 17/65 (26.2%) patients were switched to CZP due to lack of effectiveness or adverse events. A large proportion of patients in the FAS completed CZP treatment to Week 12 of the study (60/65, 92.3%), 1 patient (1.5%) permanently withdrew CZP treatment before Week 12 due to an ADR, and 4 patients (6.2%) withdrew due to “other” reasons. “Other” reasons included the patient not attending the visit, patient decision, hospitalization, and temporary interruption of CZP due to vertigo. Among the 61 patients with a Week 16 visit, 56 patients (91.8%) opted to continue CZP treatment, and 1 patient (1.6%) withdrew due to an ADR, 1 patient (1.6%) withdrew due to ineffectiveness and 3 patients (4.9%) withdrew for “other” reasons. The mean duration of RA since diagnosis (SD) for enrolled patients was 7.8 (9.8) years. The mean age of patients was similar between the two study populations, 53.8 years (SS) and 53.7 years (FAS) (Table 1). The gender of patients within the study population was predominantly female (SS: 74.0% and FAS: 73.8%) (Table 1). Disease activity and patient-reported outcomes (PROs) were similar between the SS and the FAS (Table 1). The SS had a greater median CRP (1.83mg/dL) than the FAS (1.65mg/dL) but a lower median ESR (SS: 31.0mm/h, FAS: 34.0mm/h) (Table 1). US outcomes were similar between patients in the SS and the FAS (Table 1) at baseline. At baseline, 57/65 (87.7%) patients in the FAS (69/77 [89.6%] patients in the SS) were taking concomitant DMARDs (Table 1). At Week 12, 10 (15.4%) patients in the FAS were taking concomitant DMARDs, with 4 (6.2%) patients receiving a higher DMARD dose compared to baseline.
Patient baseline characteristics.
| Full analysis set, N=65Mean (SD) [n], unless otherwise stated | Safety set, N=77Mean (SD) [n], unless otherwise stated | |
|---|---|---|
| Age, years | 53.7 (12.8) [64] | 53.8 (12.7) [76] |
| Female, n (%) | 48 (73.8) | 57 (74.0) |
| BMI, kg/m2 | 26.6 (5.0) [64] | 26.7 (5.3) [76] |
| HAQ-DI | 1.4 (0.8) [64] | 1.4 (0.8) [76] |
| DAS28 (ESR) | 5.8 (1.1) [65] | 5.6 (1.3) [76] |
| PtAAP-VASa | 66.8 (22.1) [63] | 66.0 (23.8) [74] |
| SF-36 PCS | 32.2 (7.7) [63] | 32.6 (7.6) [75] |
| SF-36 MCS | 42.4 (12.8) [63] | 41.7 (12.6) [75] |
| ESR, median mm/h (min; max)b | 34.0 (4.0; 140.0) [65] | 31.0 (4.0; 140.0) [77] |
| CRP, median mg/dL (min; max)b | 1.65 (0.0; 14.5) [64] | 1.83 (0.0; 17.0) [76] |
| Synovial hypertrophy index | 19.3 (10.9) [63] | 18.3 (10.8) [73] |
| Effusion index | 14.3 (10.6) [63] | 13.8 (10.2) [73] |
| Power Doppler signal index | 8.9 (7.0) [63] | 8.4 (6.9) [73] |
| Concomitant DMARDs,cn (%) | 57 (87.7) | 69 (89.6) |
| Sulfasalazine | 2 (3.1) | 3 (3.9) |
| Leflunomide | 10 (15.4) | 15 (19.5) |
| Hydroxychloroquine | 5 (7.7) | 8 (10.4) |
| Indometacin | 1 (1.5) | 4 (5.2) |
| Methotrexate | 45 (69.2) | 55 (71.4) |
| Prednisone | 1 (1.5) | 30 (39.0) |
| Prior anti-TNF treatment, n (%) | 17 (26.2) | 20 (26.0) |
The full analysis set was defined as all patients who had a least one dose of CZP during the study, at least one valid baseline and post-baseline effectiveness assessment, and had no important protocol deviations. The safety set consisted of all patients that took at least one dose of CZP at any point during the study.
Patient's assessment of arthritis pain was recorded on a visual analog scale (VAS) ranging from 0mm (no pain) to 100mm (maximum pain).
Patients receiving any concomitant disease modifying anti-rheumatic drugs. BMI: body mass index, CRP: C-reactive protein, DAS28: 28-joint disease activity score, DMARDs: disease modifying anti-rheumatic drugs, ESR: erythrocyte sedimentation rate, HAQ-DI: health assessment questionnaire disability index, MCS: mental component summary, NR: not reported, PCS: physical component summary, PtAAP: patient's assessment of arthritis pain, RF: rheumatoid factor, SD: standard deviation, SF-36: Short-Form 36-item Health Survey; TNF: tumor necrosis factor, VAS: visual analog scale.
Rapid improvements in clinical and PROs were observed between baseline and Week 12 (Fig. 1). An overall significant mean (SD) reduction of −0.6 (0.6) in HAQ-DI from baseline to Week 12 was achieved (p<0.001, Fig. 1A) and 33/65 (50.8%) patients achieved MCID in HAQ-DI at Week 12. A similar significant improvement was observed for DAS28 (ESR), with an overall mean reduction of −2.2 (1.5) from baseline to Week 12 (p<0.001, Fig. 1B). PROs also showed a significant improvement between baseline and Week 12, with a substantial reduction in PtAAP (VAS Scale 0–100) from 66.8 to 27.6 (p<0.001, Fig. 1C). Further to this an overall improvement in mean SF-36 PCS and SF-36 MCS was seen to rise from 32.2 at baseline to 38.4 by Week 12 (Fig. 1D) and from 42.4 at baseline to 50.1 at Week 12 (Fig. 1E) (both p<0.001), respectively.

Clinical and patient-reported outcomes at baseline and Week 12. Data are presented for the full analysis set. Values are means and error bars represent the 95% confidence intervals. ***Statistically significant difference between baseline and Week 12 mean values; p<0.001. Patient's assessment of arthritis pain (PtAAP) was recorded on a visual analog scale (VAS) ranging from 0mm (no pain) to 100mm (maximum pain). DAS28: 28-joint count Disease Activity Score, ESR: erythrocyte sedimentation rate, HAQ-DI: health assessment questionnaire-disability index, MCS: mental component summary, PCS: physical component summary, PtAAP: patient's assessment of arthritis associated pain, VAS: visual analog scale.
A comparison between anti-TNF naïve and anti-TNF first switch patient's HAQ-DI score indicated that functional improvements were seen in both anti-TNF naïve and first switch patients, although the patient numbers in these sub-group analyses were low (Fig. 2). The improvement seen in the naïve group (34 patients) was −0.67 (95% CI at Week 12: −0.86, −0.47) whereas the improvement in the first switch patients (13 patients) was numerically smaller, −0.28 (95% CI at Week 12: −0.65, 0.09). At Week 12, 27/34 anti-TNF naïve patients achieved MCID in HAQ-DI, compared to 6/13 first switch patients.
. Data are presented for the full analysis set (n=65) and patients are grouped by prior anti-TNF treatment (naïve or first-switch). Values are means and the error bars represent the 95% confidence intervals. For one patient within the first switch group the HAQ-DI score was not available at baseline. The Week 12 data are reported for the number of patients with HAQ-DI scores available at this time point.")
Mean HAQ-DI in anti-TNF naïve and first switch patients at Week 12 versus baseline (observed data). Data are presented for the full analysis set (n=65) and patients are grouped by prior anti-TNF treatment (naïve or first-switch). Values are means and the error bars represent the 95% confidence intervals. For one patient within the first switch group the HAQ-DI score was not available at baseline. The Week 12 data are reported for the number of patients with HAQ-DI scores available at this time point.
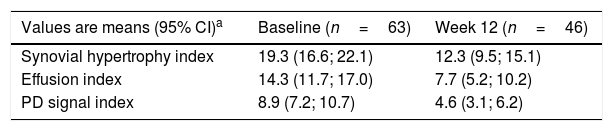
Reductions from baseline to Week 12 were seen in synovial hypertrophy index, from 19.3 to 12.3, effusion index, from 14.3 to 7.7, and PD signal index, from 8.9 to 4.6 (Table 2). These reductions were observed in both anti-TNF naïve and first switch patients (Fig. 3). The mean [95% CI] effusion index for the naïve (13.5 [10.7, 16.3]) and first switch patients (16.8 [9.4, 24.1]) differed at baseline (Fig. 3D).
Ultrasound outcomes at baseline and Week 12.
| Values are means (95% CI)a | Baseline (n=63) | Week 12 (n=46) |
|---|---|---|
| Synovial hypertrophy index | 19.3 (16.6; 22.1) | 12.3 (9.5; 15.1) |
| Effusion index | 14.3 (11.7; 17.0) | 7.7 (5.2; 10.2) |
| PD signal index | 8.9 (7.2; 10.7) | 4.6 (3.1; 6.2) |
Data are reported for the full analysis set, which was defined as the number of patients that received at least one dose of CZP and that provided at least one valid baseline and post-baseline effectiveness assessment and who had no important protocol deviations.
 synovial hypertrophy index, (B) effusion index, and (C) Power Doppler signal index. Data are reported for the full analysis set. Values are means and the error bars represent the 95% confidence intervals.")
Power Doppler ultrasound scores at baseline and Week 12 for anti-TNF naïve and first-switch patients for (A) synovial hypertrophy index, (B) effusion index, and (C) Power Doppler signal index. Data are reported for the full analysis set. Values are means and the error bars represent the 95% confidence intervals.
The safety profile of CZP was consistent with other reports of CZP in RA; no new safety signals were identified (Table 3). A total of 19 TEADRs were reported by 13/77 (16.9%) patients, 9 of which led to the discontinuation of 8/77 (10.4%) patients from the study. A total of 4 serious TEADRs were experienced by 3/77 (3.9%) patients. The serious TEADRs were lymphadenopathy mediastinal, tuberculosis, skin reaction and non-Hodgkin's lymphoma. The case of active tuberculosis was reported by the investigators to be related to CZP and the patient permanently discontinued CZP treatment. As the patient recovered after treatment with anti-tuberculosis therapy, with the disappearance of pulmonary nodules, the case was diagnosed clinically as tuberculosis, despite the absence of microbiological confirmation. The skin reaction was considered to be a medically important reaction and the patient permanently discontinued CZP treatment; the patient recovered from this event. Non-Hodgkin's lymphoma was reported 69 days after initiation of CZP in one patient, and was considered severe in intensity. The patient permanently discontinued CZP treatment due to non-Hodgkin's lymphoma. The event resulted in the death of the patient 64 days after onset. Because the study investigator stated that they were unable to confirm the relationship, UCB Pharmacovigilance Department, per protocol, classified the relationship of this adverse drug reaction conservatively as ‘related to study drug’.
Summary of treatment-emergent adverse drug reactions.
| TEADR | Safety set, N=77 |
|---|---|
| Preferred term | n (%) [# of events] |
| Any TEADRs | 13 (16.9) [19] |
| Alopecia | 1 (1.3) [1] |
| Hyperhidrosis | 1 (1.3) [1] |
| Papule | 1 (1.3) [1] |
| Rash | 1 (1.3) [1] |
| Skin reaction | 2 (2.6) [2] |
| Urticaria | 1 (1.3) [1] |
| Escherichia urinary tract infection | 1 (1.3) [1] |
| Respiratory tract infection | 1 (1.3) [1] |
| Tuberculosis | 1 (1.3) [1] |
| Conjunctivitis | 1 (1.3) [1] |
| Vision blurred | 1 (1.3) [1] |
| Lymphadenopathy mediastinal | 1 (1.3) [1] |
| Gingival ulceration | 1 (1.3) [1] |
| Pyrexia | 1 (1.3) [1] |
| Non-Hodgkin's lymphoma | 1 (1.3) [1] |
| Dizziness | 1 (1.3) [1] |
| Bronchial hyperactivity | 1 (1.3) [1] |
| Vasculitis | 1 (1.3) [1] |
| Serious TEADRs | 3 (3.9) [4] |
| Lymphadenopathy mediastinal | 1 (1.3) [1] |
| Tuberculosis | 1 (1.3) [1] |
| Non-Hodgkin's lymphoma | 1 (1.3) [1] |
| Skin reaction | 1 (1.3) [1] |
| Discontinuations due to TEADRs | 8 (10.4) [9] |
| Bronchial hyperreactivity | 1 (1.3) [1] |
| Respiratory tract infection | 1 (1.3) [1] |
| Tuberculosis | 1 (1.3) [1] |
| Skin reaction | 2 (2.6) [2] |
| Lymphadenopathy mediastinal | 1 (1.3) [1] |
| Gingival ulceration | 1 (1.3) [1] |
| Non-Hodgkin's lymphoma | 1 (1.3) [1] |
| Vasculitis | 1 (1.3) [1] |
| Death | 1 (1.3) [1] |
Data are reported for the safety set, which was defined as all patients that received at least one dose of CZP at any time during the study. Values are based on MedDRA preferred term. n=number of patients reporting at least 1 TEADR within the preferred term. TEADR: treatment emergent adverse drug reaction.
Treatment with anti-TNFs has been found to routinely manage patients’ RA symptoms. In the present 12-week, observational study, treatment with CZP led to significant improvements of both clinical outcomes and PROs. Similar improvements in clinical outcomes, in patients with RA, have been observed after 12 weeks, and up to 5 years of anti-TNF treatment, using data obtained from the Dutch and German biologic registers.22,23 However, as these observational studies were not conducted in Spain, these data are not directly comparable to the data from the present study. The recent preliminary findings from the IMPULSAR study, conducted in Barcelona, highlighted the value of US as a complimentary tool for treatment decisions.24 These findings are complimented by the observations that US outcomes, but not clinical remission (measured by DAS28<2.6, SDAI<3.3, and CDAI<2.8), were associated with prediction of 1-year X-ray progression of RA, and failure of biologic tapering in patients with RA.25,26
The clinical findings of the present study aligned with a reduction in synovial hypertrophy, effusion and PD signal indices from baseline to Week 12, as determined by PDUS, both in the present study and from Week 8 to Week 52 in the CZP-SPEED trial.27 Other anti-TNFs such as etanercept and adalimumab have also reported improvements in PDUS outcomes, which often accompany improvements in PROs.28–30 The clinical definition of RA remission is a DAS28 (ESR) <1.6 for at least 6 months, and low disease activity (LDA) is defined as DAS28 (ESR) <2.4 but >1.6 for at least 6 months, however, PDUS outcomes have identified that patients with RA, with apparent clinical remission, can still experience joint damage progression.31,32
Observational studies have shown that many patients discontinue anti-TNF therapy for various reasons,33 but patients discontinuing due to an initial lack of effect, or subsequent loss of effectiveness are less likely to achieve low disease activity or remission upon successive therapy, and are more likely to experience flares.33,34 However, in the present study, improvements in the HAQ-DI score of both anti-TNF naïve and first switch patients after 12 weeks of CZP treatment were observed, though patients who had never been treated with an anti-TNF showed a slightly stronger improvement. The number of patients in both subgroups was low, especially in the first-switch patient group (n=16), and this is likely the reason of a relatively large numerical difference for HAQ-DI being observed in this subgroup. Similarly, in the REALISTIC trial investigating the effect of CZP on clinical outcomes and PROs in a similar mixed population of naïve and switch RA patients, patients experienced comparable improvements in HAQ-DI, DAS28 (ESR) and PtAAP-VAS during the first 12 weeks of the 28-week study.4
The small number of patients enrolled in this study was a major limitation, however, 60/65 (92.3%) patients completed the treatment for the duration of the study (Week 12), and 56/61 (91.8%) of the patients that attended the visit at Week 16 opted to carry on with the treatment following the end of the study, providing data at Week 16. Nevertheless, the differences observed in primary analysis were larger than the minimally clinically significant change defined for the sample size calculation, thus the smaller population did not prevent the study from finding clinically significant differences. Another potential limitation, was that ultrasonographers were based at individual sites due to the multicenter nature of the study, which could have introduced variability between readers. However, to ensure consistency, all ultrasonographers were experts (>5 years of experience), used appropriate equipment and followed predefined recommendations to align the definition of scanning method and standardize scoring.
One case of active tuberculosis was reported during this study, this was considered to be related to CZP, and the patient was permanently withdrawn from CZP treatment. The patient provided a negative Mantoux test result prior to receiving CZP, the positive Mantoux test result was reported 222 days after receiving CZP. One death was reported, as a result of non-Hodgkin's lymphoma, and the patient was permanently withdrawn from CZP treatment prior to their death. No new safety signals were identified within the present study and the safety profile of CZP was comparable to previous trials, including the REALISTIC trial which reported incidence of TEAEs up to Week 12, in a mixed population of patients with active RA, from multiple geographic locations.4 This therefore provides further evidence that CZP has an acceptable safety profile in patients with active RA.
In this observational study in Spain, patients with RA demonstrated improvements in clinical outcomes, PROs and PDUS outcomes over 12 weeks of CZP treatment. Improvements were observed regardless of whether patients had been previously treated with an anti-TNF, although the patient numbers in these sub-group analyses were low. The safety profile was consistent with previous CZP studies, with no new safety signals identified. These results from observational clinical practice add to the findings of previous clinical studies which demonstrate the impact of CZP on signs and symptoms of RA.
FundingUCB sponsored the study and the development of the manuscript/publication and reviewed the text to ensure that from UCB perspective, the data presented in the publication are scientifically, technically and medically supportable, that they do not contain any information that has the potential to damage the intellectual property of UCB, and that the publication complies with applicable laws, regulations, guidelines and good industry practice. The authors approved the final version to be published after critically revising the manuscript/publication for important intellectual content.
This study and all costs associated with the development of this manuscript were funded by UCB Pharma.
Conflict of interestJD and PT are employees of UCB Pharma, and EN has received speaker fees from Abbvie, Roche, BMS, Pfizer, UC, Lilly, Novartis, Janssen, and Celgene GmbH.
Data availabilityQualified researchers whose proposed use of the data has been approved by an independent review panel will be given access to anonymized individual participant data and redacted study documents. Additional information is available at www.clinicalstudydatarequest.com
The authors thank the patients, the investigators and their teams who took part in this study. The authors also acknowledge Susanne Wiegratz, UCB Pharma GmbH, Monheim, Germany, for publication coordination and Maria Haughton, CMPP and Sarah Jayne Clements, PhD at Costello Medical, Cambridge, UK, for medical writing and editorial assistance in preparing this manuscript for publication, based on the authors’ input and direction.


