


Idiopathic inflammatory myopathies are a heterogeneous group of potentially treatable myopathies. They are classified, on the basis of clinical and histopathological features, into four subtypes: dermatomyositis, polymyositis, necrotising autoimmune myositis and inclusion-body myositis. Myositis-associated antibodies and myositis-specific autoantibodies are frequently found in patients with idiopathic inflammatory myopathies, and are useful in the diagnosis and classification. Anti-histidyl transfer RNA synthetase antibody is the most widely prevalent and is highly specific for polymyositis. Signal recognition particle antibody is also a specific autoantibody for polymyositis, but it is infrequent and rarely found in patients having other myositis-specific autoantibodies.
We present a man with polymyositis who had both antibodies in serum, which is considered an extremely rare clinical situation. Here we analyse the clinical course and findings, and examine the effect of the coexistence and possible interaction on prognosis.
Las miopatías inflamatorias idiopáticas son un grupo heterogéneo de miopatías potencialmente tratables. Se clasifican en 4 subtipos: dermatomiositis, polimiositis, miositis autoinmune necrosante y miositis por cuerpos de inclusión, en función de las características clínicas e histológicas. Los anticuerpos asociados a miositis y los autoanticuerpos específicos de miositis se encuentran frecuentemente en pacientes con miopatías inflamatorias, siendo útiles en el diagnóstico y clasificación. El anticuerpo anti-histidil tRNA sintetasa es el más prevalente y el más específico para polimiositis. El anticuerpo de partícula de reconocimiento de señal es también un autoanticuerpo especıfico para polimiositis, pero más infrecuente, y raramente se encuentra en pacientes que presentan otros autoanticuerpos específicos para miositis.
En este trabajo se presenta un paciente con polimiositis en el que coexisten los 2 autoanticuerpos en el suero, lo que se considera una situación clínica extremadamente rara. Aquí analizamos la evolución clínica y hallazgos para examinar el efecto de la coexistencia y la posible interacción sobre el pronóstico.
Idiopathic inflammatory myopathies (IIM) are myopathies that are potentially treatable, and they are classified as: dermatomyositis, polymyositis (PM), necrotising autoimmune myositis and inclusion body myositis. Specific antibodies for myositis are frequent in patients with IIM, and they are useful in diagnosis and classification. The anti-Jo1 antibody is the most common, and together with the anti-signal recognition particle (anti-SRP) they are the most specific for PM. Anti-SRP is rare and appears isolated in myositis.
We present an extremely rare clinical case in which anti-Jo-1 and anti-SRP coexist in the serum. We analyse the clinical evolution of the case to examine the effect of this coexistence and the possible interaction of both antibodies in the prognosis.
Clinical caseA 60 year-old man, a smoker (40packs/year), dyslipidemic hypertension and dyslipidemia, treated with 50mg/24h atenolol, 100mg/24h acetylsalicylic acid, 10mg/24h enalapril, 10mg/24h ezetimibe and 20mg/24h simvastatin (from September 2011 to February 2012; suspended due to the diagnosis of PM).
When the patient was studied he had constitutional syndrome that had developed over 2 months, with evening fever (38°C) that went into remission with paracetamol; asthenia, hyporexia and weight loss (10kg). He described proximal weakness, nocturnal dry non-productive cough and self-limiting transitory arthromyalgias, with no signs of the Raynaud phenomenon or skin disorders.
Pulmonary auscultation detected fine crackling rales in the bases and major axial muscle weakness and proximal weakness in the limbs.
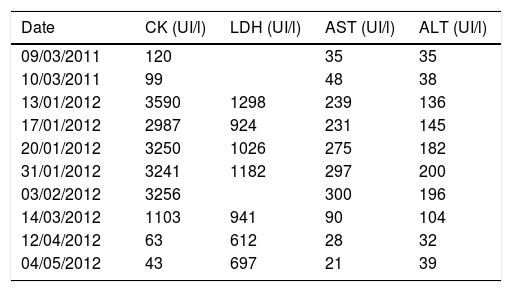
Laboratory tests showed high serum levels of creatine kinase, dehydrogenase lactate, alanine aminotransferase, aspartate aminotransferase (Table 1), reactive C protein (79.2mg/l), erythrocyte sedimentation rate (37mm/h) and leukocytosis (13,200U/mm3; 76.5% neutrophils), with normal TSH (1.90μUI/ml). Only the tumour markers Ca 15.3 (54.8U/ml; normal: 2.0–37.0) and β2-microglobulin (4.63mg/l; normal ≤3.0) were raised, although the absence of a tumour was checked by computed tomography of the thorax and abdomen. The PPD test was negative.
Evolution of serum levels of CK, LDH, AST and ALT.
| Date | CK (UI/l) | LDH (UI/l) | AST (UI/l) | ALT (UI/l) |
|---|---|---|---|---|
| 09/03/2011 | 120 | 35 | 35 | |
| 10/03/2011 | 99 | 48 | 38 | |
| 13/01/2012 | 3590 | 1298 | 239 | 136 |
| 17/01/2012 | 2987 | 924 | 231 | 145 |
| 20/01/2012 | 3250 | 1026 | 275 | 182 |
| 31/01/2012 | 3241 | 1182 | 297 | 200 |
| 03/02/2012 | 3256 | 300 | 196 | |
| 14/03/2012 | 1103 | 941 | 90 | 104 |
| 12/04/2012 | 63 | 612 | 28 | 32 |
| 04/05/2012 | 43 | 697 | 21 | 39 |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; CK: creatine kinase; LDH: dehydrogenase lactate.
Anticellular antibodies were detected in the serum. The immunoblot polymyositis profile (anti-Mi2, anti-Ku, anti Pm/SCL, anti-Jo1, anti-PL12, anti-PI7, anti-SSA 52kDa, anti-EJ, anti-OJ and anti-SRP) was positive for anti-Jo-1 and anti-SRP.
Haplotypes for celiac disease were analysed; the patient was a heterozygote carrier of the DRB1*03:01-DQB1*02:01-DQA1*05:01 haplotype, although the anti-transglutaminase IgG and IgA antibody titrations were negative.
The computed tomography results are shown in Fig. 1, while the bronchoscopy study only showed supraglottal polypoidal lesions. Bronchial aspiration cytology was negative for neoplastic cells.
. (A) ACT of the thorax at diagnosis revealed the presence of discrete interstitial infiltrates in ground glass opacity in the bases and periphery of the lungs, and to a lesser degree, in the mid-fields, with multiple mediastinal lymphatic ganglia of less than 17mm and segmental atelectasis of the lower right lobe. (B) At 3 months the angio ACT showed no images of pulmonary thromboembolism, although extensive bilateral involvement of the lungs was observed, with interstitial infiltrates with ground glass opacity and interlobular septal thickening, as well as areas of fibrosis, while the anterior pulmonary fields were less involved.")
Evolution of the pulmonary involvement shown by axial computed tomography (ACT). (A) ACT of the thorax at diagnosis revealed the presence of discrete interstitial infiltrates in ground glass opacity in the bases and periphery of the lungs, and to a lesser degree, in the mid-fields, with multiple mediastinal lymphatic ganglia of less than 17mm and segmental atelectasis of the lower right lobe. (B) At 3 months the angio ACT showed no images of pulmonary thromboembolism, although extensive bilateral involvement of the lungs was observed, with interstitial infiltrates with ground glass opacity and interlobular septal thickening, as well as areas of fibrosis, while the anterior pulmonary fields were less involved.
The electromyogram showed a predominantly proximal myopathic process, with the presence of discrete signs of inflammatory activity that may appear in polymyositis.
Quadriceps biopsy showed discrete distortion of the fascicular architecture with occasional atrophic perifascicular fibres. Occasional fibres were observed within the fascicules, with nuclear centralisation and signs of regeneration. The presence of perimysial and endomysial inflammatory lymphomonocytic infiltrates stood out.
The diagnosis is based on raised levels of muscle enzymes, muscle biopsy, myositis-specific antibodies and anomalies in the electromyogram.
Given these findings, treatment commenced with oral prednisone, 80mg/24h (1mg/kg/24h). The muscle pain and fever remitted quickly and muscle enzymes normalised after 3 months (Table 1). However, the patient had an episode of deep vein thrombosis and progressive dyspnoea, even when resting, accompanied by a non-productive cough. Cyanosis was detected in the acral areas, with an O2 saturation of 92% with the oxygen therapy mask set to high flow.
Venous Doppler ultrasound scan detected a superficial occlusive thrombus in the femoral vein, in the popliteal vein and in the origin of the tibial peroneal trunk. A clinical improvement was achieved using intravenous anticoagulant therapy in 3 pulses (1g/24h) of methylprednisolone and cyclophosphamide.
DiscussionAnti-Jo-1 antibodies are the most common in IIM (20–30%).1 They are associated with the anti-synthetase syndrome, which is characterised by polymyositis, diffuse interstitial pulmonary disease, polyarthritis, Raynaud phenomenon and hyperkeratosic skin lesions, with erythema over the metacarpophalangeal and interphalangeal joints.2 Patients with anti-Jo1 have myopathy (90%) and diffuse interstitial pulmonary disease (70%),3 and the titration may be correlated with disease activity.4
Anti-SRP antibodies recognise the cytoplasmatic protein that binds to synthesised protein signal sequences for translocation to the endoplasmic reticule. They are found in 4% of adults with polymyositis and are not usually associated with other myositis autoantibodies.5 They may trigger acute polymyositis, with severe muscle weakness, cardiac involvement, swift progression, a poor immunosuppressor response and increased mortality.6 Muscle biopsies show abundant signs of cellular degeneration and regeneration, necrotic cells and a discrete inflammatory infiltrate. Nevertheless, the biopsy findings in our patient did not display this anti-SRP anatomo-pathological phenotype.
Clinically, anti-Jo-1 antibodies are associated with diffuse interstitial pulmonary disease, and anti-SRP antibodies are associated with the acute onset of the disease, sustained increase of creatine kinase and severe weakness of the patient,7 as was seen in this patient.
Although the paraneoplastic form of inflammatory myopathies is known, the tumour has been clinically ruled out in our patient.
Statin toxicity, which is present in 7–29% of cases, is associated with inflammatory myopathies,8 as is the finding in serum of the anti-3-hydroxy-3-methylglutaril-enzime-reductase (anti-HMGCR) antibody, the pharmacological target of the statins. Myopathic symptoms usually disappear when the treatment is interrupted, and immunosuppressor therapy is needed.9 In the case of statin-induced necrotising myopathy the symptoms have been found to persist, even when the treatment is interrupted, as occurred in our patient.
However, no necrotic or regenerative fibres were found in the muscle biopsy, although foci of lymphocytes were found, strengthening the diagnosis of PM.
The celiac disease haplotype of this patient confers the risk of autoimmune (odds ratio 3.6) and 15.5 times the risk of anti-Jo-1.10
ConclusionsThe case presented here is unusual in clinical practice. PM is a rare disease in Spain (3.9/100,000). The coexistence of both autoantibodies is considered to be extremely rare, and in this case the antibodies were also useful in the diagnosis of autoimmune myopathy and the anti-synthetase syndrome. The association with anti-SRP antibodies caused severe muscle deterioration and a poor prognosis.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Melguizo Madrid E, Fernández Riejos P, Toyos Sáenz de Miera FJ, Fernández Pérez B, González Rodríguez C. Coexistencia de anticuerpos anti-histidil-tRNA-sintetasa y anti-partícula de reconocimiento de la señal en un paciente con polimiositis. Reumatol Clin. 2019;15:e111–e113.


