



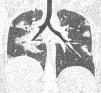
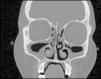
Eighteen-year-old female patient with a history of refractory asthma, recurring sinusitis, and insterstitial lung disease diagnosed in February 2021, whose current illness debuted in January 2022 with cough, wheezing, and dyspnoea. The initial physical examination revealed bilateral rales, localised dermatosis on the back of both hands characterised by pruritic erythematous-violaceous macules, which did not whiten when pressed. Laboratory tests on admission revealed 15.4 × 109/l leukocytes, with 45% eosinophilia, and elevated acute phase reactants (CRP and ESR). Paranasal sinus tomography displayed parasinus hypertrophy, chronic sinusitis. Chest CT scan without contrast evinces multilobar ground-glass images involving more than 50% of the pulmonary parenchyma. Bronchoscopy was performed; infectious causes were ruled out, and predominantly eosinophilic inflammatory cells were demonstrated. Nasal biopsy confirmed eosinophilic inflammatory infiltrate without granulomas. Histopathological studies against neutrophilic cytoplasm were negative. Eosinophilic granulomatosis with polyangiitis was diagnosed and treatment with high-dose glucocorticoids was initiated and the pulmonary and skin lesions improved. The patient was discharged with treatment based on gradual reduction of glucocorticoids (described in the PEXIVAS protocol), methotrexate 15 mg per week and mepolizumab 100 mg subcutaneous monthly. After 24 months of treatment, all symptoms have remitted and biochemical control revealed less than 1% eosinophils, normalised acute phase reactants, and a Birmingham Vasculitis Activity Score (BVAS) of 0. The patients has displayed no adverse effect during the course of treatment.
Eosinophilic granulomatosis with polyangiitis (EGPA) is an uncommon immunomediated disease. The main characteristics are late onset asthma, peripheral and tissue eosinofilia.1 The diagnostic gold standard is biopsy with evidence of eosinophilic infiltrate and fibrosing necrosis. Glucocorticoides are the cornerstone of treatment.2 However, biological treatments, such as mepolizumab, a humanised monoclonal antibody against the IL-5 receptor, has proven to be an effective treatment for EGPA.3 Classification criteria recently updated uphold the diagnosis in this case and avoid delay in treatment.1 Despite the fact that the recommended dose of mepolizumab is usually 300 mg per month, it is an expensive drug.4 This is why we originally opted to use methotrexate, prednisone, and low-dose mepolizumab in this case, which yielded a favourable outcome.4,5 (Figs. 1 and 2).


All individuals designated as authors have participated in the work and have taken public responsibility for its content.
Ethical considerationsWritten informed consent was obtained from the patient for the use of the image and the publication of the details of her case.
FundingNo funding was received.
The authors have no conflict of interests to declare.


