




The haemostatic system acts in concert with inflammation, so that after inflammatory response various mediators activate the haemostatic system through endothelial dysfunction, platelet activation and coagulation promoting thrombosis, which is termed thromboinflammation. In this process, the inflammasome acquires special relevance; its stimulation promotes innate and adaptive immune responses. Inflammasome activation plays an important physiopathological role in several disorders with inflammatory and thrombotic phenomena. The role of thromboinflammation has become relevant in the COVID-19 pandemic, in which a cytokine storm has been described as one of the mechanisms responsible.
El sistema hemostático actúa en concierto con la inflamación, de forma que tras la respuesta inflamatoria diversos mediadores activan el sistema hemostático a través de disfunción endotelial, activación plaquetar y de coagulación, promoviendo la trombosis, lo que se ha denominado tromboinflamación. En este proceso adquiere especial relevancia el inflamasoma, cuya estimulación promueve respuestas inmunes innata y adaptativa. La activación del inflamasoma juega un papel fisopatológico importante en diversas patologías que cursan con fenómenos inflamatorios y trombóticos. El papel de la tromboinflamación se ha puesto de relevancia en la pandemia por COVID-19, en la que se ha descrito una tormenta de citocinas como uno de los mecanismos responsables.
The survival of living organisms depends on their ability to produces a fast and effective response to infection, haemorrhaging and tissue damage, due to the assistance of innate defence mechanisms, such as the haemostatic system and the immune system.
The haemostatic system acts in cooperation with the inflammatory cascade creating an haemostatic-inflammatory cycle, in which each one of the processes promotes the activation of the other, with a positive feedback system. The communication between both takes places at a level of all the components of the haemostatic system, including endothelial cells, platelets, coagulation proteins, natural anticoagulation proteins, natural anticoagulant system and fibrinolytic activity. During the inflammatory response, different mediators, and particularly the cytokines play a central role impacting the haemostatic system through endothelial dysfunction, increasing platelet reactivity, activation of the coagulation cascade, reduction of the natural anticoagulant systems and suppression of fibrinolytic activity. The interaction between homeostasis and inflammation explains the prothrombotic tendency, which is known as thromboinflammation.1 During this process inflammasome acquires special relevance. This is a molecular platform which is triggered as an innate response of the organism to the presence of pathogens, whose abnormal activation leads to numerous inflammatory states and cardiovascular processes of a thrombotic nature.
InflammasomeThis is a multimeric platform consisting of a sensor protein, an adaptor protein and an effector protein which is a systeine protease called procaspase-1. The sensor protein includes NLRs [Nucleotide-binding Oligomerization Domain (NOD) and Leucine Rich-Repeat (LRR) type receptors and the adaptor molecule ASC (Apoptosis-Associated Speck-like protein). Among the diverse types of inflammasomes, the most characteristic is the NLRP3, also called cryopyrin, the gene of which is found in chromosome 1, for its participation in inflammatory processes and its expression in innate immunity cells, such as macrophages, monocytes, dendritic cells, neutrophils, lymphocytes, epithelial cells, endothelial cells and osteoclasts.2,3 These structures are involved in the recognition of pathogen-associated molecular pattern stimuli (PAMPs) or damage-associated molecular patterns (DAMPs). Following exposure to these stimuli the inflammasome forms a complex, so that the procaspase-1 is converted through an autocatalytic process into caspase-1. In turn, caspase-1 becomes interleukin 1β (IL1β) and IL18 in its active forms, leading to inflammation. The secondary inflammatory activation to DAMPs is called sterile inflammation in contrast to that induced by PAMPs.4–7
The stimulation of inflammasome by PAMPs and DAMPs triggers proinflammatory and antimicrobial events through innate and adaptative responses.8 Also, the caspases split Gasdermine D, triggering the process of pyroptosis, which includes the formation of pores in the cellular membrane (e.g. in monocytes), which leads to the generation of microparticulars rich in tissue factor (TF).9 The TF is the main activator of the haemostatic system in vivo and contributes to thrombosis, when the IX and X factors of coagulation are activated and promotes the generation of thrombin which converts fibrinogen into fibrin, which is the most important structural component of the thrombus. Finally, inflammasome induces an adaptative response mediated by T lymphocytes through the activation of the Toll-like (TLRs) receptors and concourse of the transcription factor NFkB. As a result, release of the pro-inflammatory cytokines occurs which will also trigger thrombotic processes (Fig. 1).10 On an experimental level it has been demonstrated that inflammasome activation (NLRP3) and the releases of IL1β in mice deficient in CD39, (an ectonueleotidase whose deficiency is associated with immune diseases), showed an increase in the expression of TF, fibrin and the formation of extracellular traps of neutrophils (NETs) indicating the participation of innate immunity and the creation of a prothrombotic environment.11
Mechanisms through which inflammation induces haemostatic change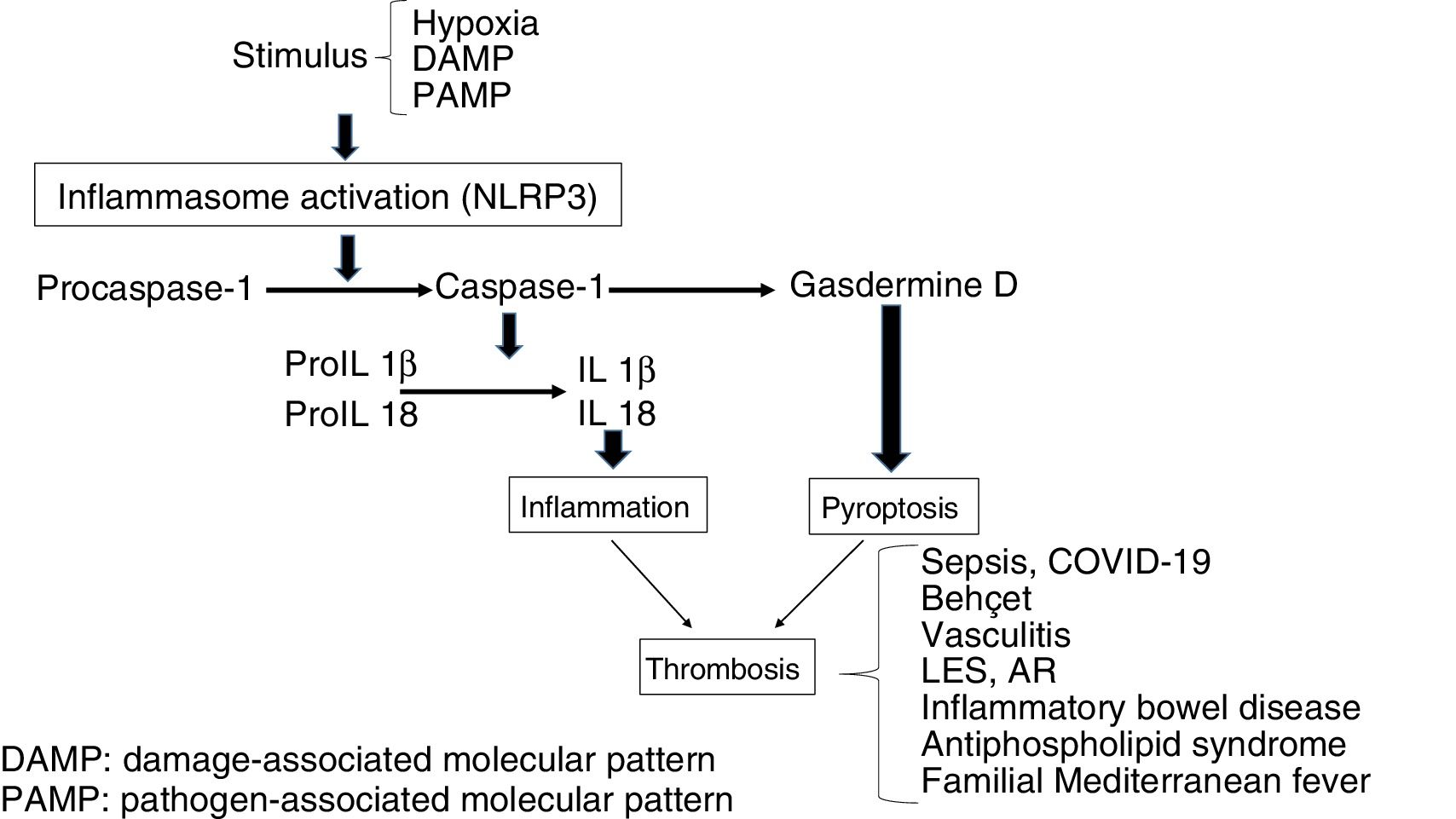
Regardless of aetiology, inflammation helps to alter the balance between procoagulant properties and vascular endothelium which acquires a pro-thrombotic phenotype. Once activated, the endothelial cells secrete procoagulating and antifibrinolytic factors, such as TF, Von Willebrand factor, thromboxane A2 and tissue plasminogen activator inhibitor (PAI-1). When vascular integrity is lost, the TF is exposed, which binds to factor VII and initiates activation of the coagulation in vivo with the generation of thrombine that converts fibrinogen into fibrin. Also, the endothelial activation involves an increase in adhesion molecules such as VCAM-1 and ICAM-1 which play a major role in the interaction of the neutrophils and platelets and in the release of proinflammatory cytokines like IL1, IL6 and TNF-α, which will also mediate the procoagulant actions of the endothelium. Finally, endothelial activation interferes in the function of the natural anticoagulating systems, such as the C protein system and the tissue factor protein inhibition (TFPI), leading to a pro-thrombotic environment. Inflammation also involves platelet activation with the release of pro-coagulating substances and proinflammatory cytokines which promote a procoagulant state.12–14
Mechanisms through which haemostatic activation promotes the inflammatory responseCommunication between inflammation and haemostasis is two-directional. Individual components of the haemostatic system, such as the Xa factor or the TF/VIIa complex are involved in the inflammatory response through the production of inflammatory mediators through endothelial cells, leukocytes and platelets. A major mechanism through which the coagulation factors increase the inflammatory response is through the binding to receptors activated by proteases or PARs. The PARs are a family of receptors with four members, PAR-1 to PAR-4, which are located in endothelial cells, leukocytes, platelets, fibroblasts and smooth muscle cells and which, after protheolytic activation by the Xa factor or the TF/VIIa complex, an inflammatory response occurs with the release of cytokines, chemokines, adhesive molecules and growth factors. Consequently, PARs play an essential role in the relationship between inflammation and haemostasis.12–14
Prothrombotic/proinflammatory states associated with inflammasomeThe activation of inflammasome plays a major physiopathological role in different clinical situations in which there is interaction between coagulation and inflammation which contributes to a prothrombotic phenotype.15
Infection by COVID-19The current pandemic induced by the coronavirus COVID-19 is a good example of viral infection associated with a systemic inflammatory response and activation of coagulation in symptomatic patients. However, as has been peevishly pointed out, disseminated intravascular coagulation (DIC) is a recognised complication of bacterial infections; coronavirus infection may also cause it and condition thrombotic phenomena in diverse regions. Although the mechanism of coagulopathy has not specifically established, it is known that viral infections induce a systemic inflammatory response accompanied by a “storm of cytokines”, which alter the balance between pro and anti coagulant mechanisms and therefore promote endothelial impairment, raising of the levels of von Willebrand factor and tissue factor, promoting activation of coagulation mechanism. Changes in coagulation and thrombotic complications are common in these patients. In a retrospective series of 183 patients Tang et al. reported that 71.4% of those who died met with the DIC criteria, compared with .6% of the survivors. 16 In this, and in other series, alterations of coagulation tests have been described, including the increase of dimer D (36%–50%), prolongation of prothrombin time (30%) and partially activated thromboplastine (PATP) (16%) and thrombocytopenia (0%–30%). The patients infected by this virus, in addition to developing DIC, may present with venous thrombosis and/or pulmonary embolism, and arterial embolism, with episodes of ischemia having been described in the lower limb toes which may lead to gangrene. Very recent results obtained from patients in the area of Wuhan in China have demonstrated dimer D, a marker of thrombin generation and of fibrinolysis, which constitutes a relevant prognostic indicator of mortality. These studies indicate that dimer D levels above 1000 ng/mL s are associated with an 18 times higher risk of mortality, to the point where today this is included the screening of all positive symptomatic COVID -19 patients. The fact that there is a coagulopathy present in these patients has led to promoting antithrombotic strategies, especially in patients who are admitted to the ICU and/or show organic damage or have had ischemic episodes as previously described. Although the best antithrombotic strategy has yet to be established, it appears that low weight molecular heparins at prophylactic or intermediate doses should be administered to these patients after their admission to the ICU or when dimer D values are 4 time higher than normal.16–18
SepsisThis is a clinical syndrome characterised by an exaggerated response of the host to the infection, which leads to an uncontrolled inflammatory response and generalised activation of the haemostatic system. After exposure to pathogens, toxins, microbials or PAMPs, the endothelial cells acquire a proapoptotic proinflammatory and prothrombotic phenotype. They also damage the glycocalix and impair vascular tone. A common complication of patients with sepsis is DIC, which is characterised by general activation of coagulation through the TF in response to proinflammatory cytokines. Also, this leads to a drop in the function of natural anticoagulants and suppression of fibrinolysis through an increase in PAI-1. The result is generalised microthrombosis which leads to organ damage and high mortality. Multiple evidence exists that inflammasome is involved (NLRP3) in the pathogenesis of sepsis, with an effect on the generation of IL1β, mitochondrial response, and for its cardiovascular, renal and central nervous system effects.19–21
Behcet diseaseThis is a multisystemic vasculitis which causes oral and genital ulcers, skin wounds, arthritis, uveitis, gastrointestinal ulcers and venous and arterial thrombotic manifestations in 12% of cases, with those affecting the venous region being the most frequent. The participation of neutrophils and the generation of NETs has been linked to thrombotic risk. An increase in NLRP3 and IL1β22,23 expression has also been observed.
Positive ANCA vasculitisVenous thromboembolism is a common complication in patients with antineutrophil auto-antibodies (ANCA). An increase in TF and procoagulant microparticles was observed which contributed to the hypercoagulability. There is also an increase in cytokines, including IL1β y TNF-α, which lead to an increase in TF responsible for thrombosis, and a raised expression of NLRP3, NLRC5, IL1β and IL18.24,25
Inflammatory bowel diseaseTogether with Crohn’s disease and ulcerous colitis this is associated with an increased risk of venous and arterial thromboembolism, possibly related to cellular and molecular changes involved in the haemostatic process. This risk has been described as being 2–3 times higher than that of the general population, exacerbating during periods of activity and lowering during periods of remission. Inflammatory bowel diseases are also a risk factor for splanchnic, portal and mesenteric thrombosis and are associated with increased risk of myocardial infarction and stroke. Among the proposed mechanisms an increase in inflammatoy cytokines has been described which are involved in a prothrombotic state manifested as thromocytosis, raised TF, II, V, VII, VII and X and fibrinolysis deficiency. The frequent use of corticoids can exacerbate the prothrombotic state, to which other factors are added, including malnutrition and the insertion of central venous lines. A raised expression of NLRP3 and IL1β has been observed, together with a lowered expression of NLRP6 and NLRP12.26,27
Systemic erythematosus lupus (SEL) and rheumatoid arthritis (RA)Different autoimmune diseases, including SEL and RA are considered to be separate risk factors for the development of venous and arterial thrombosis, which play a part in increasing mortality (up to 25%) in these patients, that is partly linked with the presence of antiphospholipid antibodies.28,29 Furthermore, cardiovascular diseases of an atherosclerotic nature are a major cause of morbimortality, which has been related to immune deregulation, hyperhomocysteinaemia, inflammation and endothelial dysfunction, with expression of adhesion molecules (ICAM-1 and VCAM-1) and procoagulating factors. In both entities a raised expression of NLRP3 has been described, and an increase in caspase-1 e IL1β and IL18.30,31 Also, polymorphisms in NLRP1 have been associated with these diseases.32,33 Apoptotic deregulation is also present in patients with rheumatic disease, in such a way that in inflammatory cells such as lymphocytes the apoptosis lowers through the over-expression of BCL2, inducing tissue damage with a higher exposure of antigens and production of auto-antibodies.34
Antiphospholyic syndromeThis is considered to be the most commonly acquired thrombophilic state, defined by thrombosis or gestational morbidity associated with the presence of antiphospholipid antibodies. Venous thrombosis at lower limb level and thrombosis in cerebral circulation are the most common locations for the presence of venous and arterial thrombosis, respectively. Antiphospholipid antibodies play a major pathogenic role, since they induce thrombosis through different mechanisms, including endothelial activation and monocytosis with raised TF and thromboxane A2. Furthermore, inflammasome activation through NLRP3 has been demonstrated, with increased caspase-1 and the production of IL1β.35,36 In antiphospholipid syndrome an increase in apoptosis has also been described as inducing the exposure of phosphatidylserine which would represent an additional mechanism to drive procoagulant activity.37
Familial Mediterranean feverThis is characterised by acute, recurrent fever, accompanied by polyserositis, with arterial and venous thrombosis being common as a consequence of endothelial lesion/impairment. The most current hypothesis is based on the pathogenic role of the gene MEFV, which codes a protein of the pyrin family. A mutation of this gene leads to an increase in the leukocyte expression of proinflammatory cytokines (IL1, IL6, IL18), which lead to endothelial damage and activate coagulation, triggering thrombosis. Activation of inflammasome, via NLRP3 has also been observed, with uncontrolled production of IL1β.38–40
To conclude, better knowledge regarding the association between thrombosis and inflammation may be of use in the determining new therapeutic strategies. Research of agents which act against inflammasome may be a new therapeutic pathway against thrombosis. For example, it has been demonstrated that a specific NLRP3, MCC950 inhibitor could be used in managing certain diseases with high inflammatory components.41,42
Reference to the influence of inflammasome in the context of thrombosis may, therefore, have a cross-sectional application for the prevention of cardiovascular and thrombotic manifestation associated with inflammatory processes.
Conflict of interestsNone.
Please cite this article as: Páramo JA. Respuesta inflamatoria en relación con COVID-19 y otros fenotipos protrombóticos. Reumatol Clin. 2022;18:1–4.


