




Primary Sjögren syndrome (PSS) is a chronic inflammatory autoimmune disease. Interstitial lung disease (ILD) can be an extraglandular complication.
ObjectiveTo evaluate the clinical characteristics of patients diagnosed with PSS with ILD.
MethodsMulticentre cohort study with 25 patients diagnosed with PSS and ILD. Data of PSS, prognostic factors, pulmonary involvement variables, complementary tests that suggest a worse diagnosis and treatment given were collected. EULAR index was measured for Sjögren's syndrome.
ResultsWe identified 25 patients. In 15/25 the diagnosis of ILD was done before the diagnosis of PSS. The histopathological patterns found were: 12 NSIP, 5 UIP, 4 OP, 2 LIP. PFRs showed restrictive pattern. The majority of the patients received glucocorticoid therapy, antimalarial or immunosuppressive treatment.
ConclusionsPatients affected with PSS must be screened to catch a precocious diagnosis of ILD. The majority of the patients were diagnosed of ILD before being diagnosed of PSS. Multicenter cohorts are increasingly demanded and a multidisciplinary management is needed.
El síndrome de Sjögren primario (SSP) es una enfermedad inflamatoria autoinmune. La enfermedad pulmonar intersticial (EPI) puede ser una complicación extraglandular.
ObjetivoEvaluar las características clínicas de los pacientes diagnosticados de SSP con EPI.
MétodosEstudio de cohortes multicéntrico con 25 pacientes diagnosticados de SSP y EPI. Se recopilaron datos propios del SSP, factores pronóstico, variables de medida de la afectación pulmonar, pruebas complementarias que sugieren un peor pronóstico, así como el tratamiento recibido. Se calculó el índice EULAR para el síndrome de Sjögren.
ResultadosSe identificaron 25 pacientes. Quince de ellos fueron diagnosticados de EPI antes que de SSP. Los patrones histopatológicos encontrados fueron 12 con neumonía intersticial inespecífica, 5 con neumonía intersticial común, 4 con neumonía organizada, 2 con neumonía intersticial linfocítica. Las pruebas de función respiratoria mostraron un patrón restrictivo. La mayoría de los pacientes recibió un tratamiento con glucocorticoides, antipalúdicos o inmunodepresores.
ConclusionesLos pacientes afectados por SSP deben ser sometidos a pruebas para detectar un diagnóstico precoz de EPI. La mayoría de los pacientes fueron diagnosticados de EPI antes que de SSP. Los estudios de cohortes multicéntricos son cada vez más demandados y se precisa una gestión multidisciplinar.
Primary Sjögren syndrome (PSS) is a chronic inflammatory autoimmune disease characterized typically by lymphocytic infiltration of the exocrine glands resulting in the sicca syndrome. Although sicca features are the central clinical manifestations of the disease, PSS can cause systemic extraglandular manifestations.1
Pulmonary manifestations can be an extraglandular complication, with reported prevalence varying widely (9–75%), depending on the methods of detection and patient selection. Lung involvement (symptoms and either pulmonary function testing or radiographic abnormalities) can occur in 10–20% of patients.2 Image techniques, such as chest radiographs and high resolution computerized tomography (HRCT), are the most useful tools to detect lung involvement. A plain chest X-ray has a low sensitivity to detect early lung involvement. Studies that systematically perform HRCTs, even in asymptomatic patients, report higher rates of pulmonary abnormalities compared with studies based on clinical symptoms.3 HRCT may identify ground-glass attenuation, thin-walled cysts, honeycombing, reticular pattern, small nodules and enlarged mediastinal lymph nodes. Several histopathologic patterns have been described in PSS, including nonspecific interstitial pneumonia (NSIP), usual interstitial pneumonia (UIP), organizing pneumonia (OP) and lymphocytic interstitial pneumonia (LIP). Most of these patterns are known as diffuse interstitial lung disease (ILD). LIP has a typical radiographic appearance in ground glass opacities with thin-walled cysts, and the presence of these cysts on HRCT scan should raise clinical suspicion for PSS with ILD.4,5
NSIP is the most commonly observed histopathological pattern in connective tissue diseases with ILD, including PSS. In patients with PSS and ILD, HRCT and histopathological findings are well correlated6 and a lung biopsy is usually not recommended. However, radiographic features suggestive of lymphoma (consolidation, large nodules or pleural effusions) require thorough investigation7 (Figs. 1–3).



Bronchiolitis and bronchiectasis are the most common airway manifestations (occasionally secondary to the fibrosis of the ILD). Patients with PSS are also at an increased risk of lymphoma. Other lung abnormalities have been described: amyloidosis, granulomatosis lung disease, pseudolymphoma, pulmonary hypertension and pleural disease.2
Patients with ILD also show a restrictive functional defect that limits respiratory functionality in a variable range of severity. Diffuse ILD is the most serious form of lung involvement due to its potentially progressive nature and simultaneously its risk of respiratory failure.8
Pulmonary function test (PFT) are also necessary to demonstrate functional respiratory disability, even in asymptomatic patients. PFT might also be done very early after disease onset, and the most reliable variable to detect functional defect related to an ILD is DLCO. The presence of lower DLCO has been associated to higher mortality rates.
Recently, an estimated prevalence of 22% of diffuse ILD in PSS in a population-based hospital patient cohort has been described. Pulmonary involvement is related to an increased mortality in PSS patients.9
ObjectivesConsidering the low prevalence of the lung involvement in PSS, the heterogeneous pattern that can be found in the lung and the severity of the ILD, the aim of this study was to evaluate the clinical characteristics of patients diagnosed with PSS with ILD at 5 Rheumatology clinics in the Barcelona area.
Materials and methodsWe performed a multicenter cohort in various Hospitals in Barcelona including 25 patients with PSS and ILD. From a total of 743 patients with PSS, 25 (3.4%) developed ILD. The average of follow up of those patients with PSS was 15 years (range 5–35).
An identical database was used in the 5 clinics to collect the information. The information of the study, included in the database, was collected retrospectively during 2013 from the 5 different clinics participating in the study.
The PSS was diagnosed according to the American-European Consensus Group (AECG) criteria for PSS from 2002. Registry data included: age at evaluation, gender, date of diagnosis of PSS, the type of criteria fulfilled for PSS, immunology data (anti-Ro 60, anti-Ro 52, anti-La, RF, ANA). We also included the poor outcome prognostic factors of the illness, such as hypergammaglobulinemia or b2-microglobulin and acute phase reactants.
Lung involvement disease variables included: date of diagnosis of ILD, results of bronchoalveolar lavage (BAL), HRCT, pulmonary biopsy if needed, PFTs with DLCO. We recorded complementary tests that suggest poor further prognosis of the outcome of the disease: echocardiography to measure pulmonary hypertension and a walking test to determine the functional respiratory capacity. We also collected clinical information: arthritis, Raynaud phenomenon, polyneuropathy, parotid gland disease, periodontal disease and purpura or cryoglobulinemia. Treatment received before and during the process of the disease were collected, such as the complications or associated processes.
Eular Sjögren's syndrome disease activity index (ESSDAI) was calculated.10 The ESSDAI is a clinical index designed to measure disease activity in patients with PSS. There are 12 domains associated with disease activity (scoring goes from 0 to 3).
The data were analyzed descriptively.
ResultsIn our cohort (n=25), the mean age at evaluation was 70 years old (±8 years). The majority were females. In 15/25 of the patients the diagnosis of ILD was done before the diagnosis of PSS. The mean disease duration at evaluation was 7±10 years. The wide variety of histopathological patterns from our cohort was: 12 NSIP, 5 UIP, 4 OP, 2 LIP. Apart from those, one patient was diagnosed of bronchoalveolitis and another one of lung fibrosis. The majority of patients (20/25) referred keratoconjunctivitis sicca, and 20/25 had xerosis. The Schirmer test was pathological in 20 patients. We did 15 salivary scintigraphies, 11 were pathological, and 15 lip biopsies were performed, 10 with the specific focal lymphocytic sialoadenitis, characterized by one or more tightly aggregated lymphocytes (over 50) adjacent to normal gland tissue. These aggregates of lymphocytes are called foci and their total density is called a “focus score”, highly specific of PSS pattern. Anti-Ro 60 was positive in 16/25 patients, anti-Ro 52 was determined positive in 4/9 patients. Anti-La was positive in 10/25 patients, RF in 17/25 and ANA in 22/25 (most of these determinations had a spotted pattern and a title ≥320). Ten patients had hypergammaglobulinemia and the determination of b2microglobulin was normal almost in every patient. The mean ESR when data was collected was 35mm/h and the PCR was 2.3mg/dL. BAL was performed in 11 patients, 5 of them with >10% of lymphocytes (5–70%). In 2 patients a lung biopsy was needed to confirm the diagnosis.
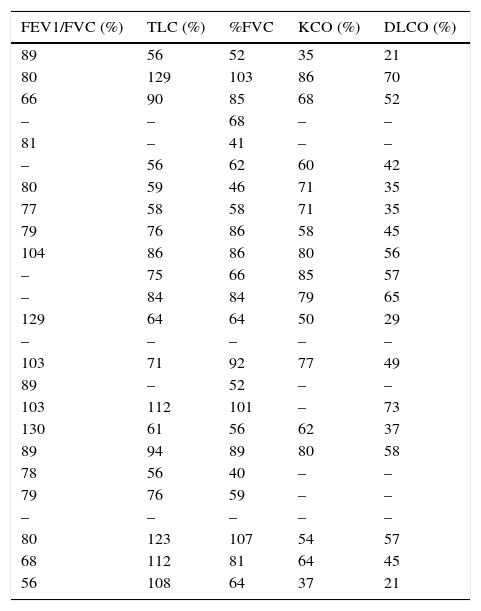
The PFRs results are shown in Table 1, the majority had a clear restrictive pattern.
Results of the PFRs.
| FEV1/FVC (%) | TLC (%) | %FVC | KCO (%) | DLCO (%) |
|---|---|---|---|---|
| 89 | 56 | 52 | 35 | 21 |
| 80 | 129 | 103 | 86 | 70 |
| 66 | 90 | 85 | 68 | 52 |
| – | – | 68 | – | – |
| 81 | – | 41 | – | – |
| – | 56 | 62 | 60 | 42 |
| 80 | 59 | 46 | 71 | 35 |
| 77 | 58 | 58 | 71 | 35 |
| 79 | 76 | 86 | 58 | 45 |
| 104 | 86 | 86 | 80 | 56 |
| – | 75 | 66 | 85 | 57 |
| – | 84 | 84 | 79 | 65 |
| 129 | 64 | 64 | 50 | 29 |
| – | – | – | – | – |
| 103 | 71 | 92 | 77 | 49 |
| 89 | – | 52 | – | – |
| 103 | 112 | 101 | – | 73 |
| 130 | 61 | 56 | 62 | 37 |
| 89 | 94 | 89 | 80 | 58 |
| 78 | 56 | 40 | – | – |
| 79 | 76 | 59 | – | – |
| – | – | – | – | – |
| 80 | 123 | 107 | 54 | 57 |
| 68 | 112 | 81 | 64 | 45 |
| 56 | 108 | 64 | 37 | 21 |
TLC: total lung capacity; FEV1: Force dexpiratory volume in 1s; FVC: forced vital capacity; DLCO: carbonmonoxide diffusing capacity; KCO: CO transfer coefficient.
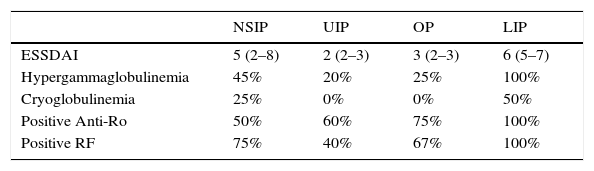
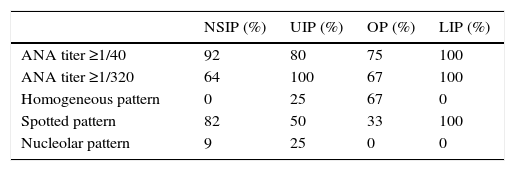
Analytical findings and ESSDAI results, as well as titers and patterns of ANA in the different pulmonary patterns are shown in Tables 2 and 3.
Analytical findings and ESSDAI results in the different pulmonary patterns.
| NSIP | UIP | OP | LIP | |
|---|---|---|---|---|
| ESSDAI | 5 (2–8) | 2 (2–3) | 3 (2–3) | 6 (5–7) |
| Hypergammaglobulinemia | 45% | 20% | 25% | 100% |
| Cryoglobulinemia | 25% | 0% | 0% | 50% |
| Positive Anti-Ro | 50% | 60% | 75% | 100% |
| Positive RF | 75% | 40% | 67% | 100% |
ESSDAI: Eular Sjögren's syndrome disease index; RF: rheumatoid factor; NSIP: nonspecific interstitial pneumonia; UIP: usual interstitial pneumonia; OP: organizing pneumonia; LIP: lymphocytic interstitial pneumonia.
Titers and patterns of ANA in the different pulmonary patterns.
| NSIP (%) | UIP (%) | OP (%) | LIP (%) | |
|---|---|---|---|---|
| ANA titer ≥1/40 | 92 | 80 | 75 | 100 |
| ANA titer ≥1/320 | 64 | 100 | 67 | 100 |
| Homogeneous pattern | 0 | 25 | 67 | 0 |
| Spotted pattern | 82 | 50 | 33 | 100 |
| Nucleolar pattern | 9 | 25 | 0 | 0 |
NSIP: nonspecific interstitial pneumonia; UIP: usual interstitial pneumonia; OP: organizing pneumonia; LIP: lymphocytic interstitial pneumonia.
Four patients had pulmonary hypertension and the walking test was performed in 20 patients, showing pathological findings in half of them. We collected 6 patients with arthritis, 5 with Raynaud phenomenon, 3 with polyneuropathy and 6 with parotid gland disease, 1 with periodontal disease, 4 with purpura or cryoglobulinemia. As with other associated processes we collected: 4 osteoporosis, 2 respiratory infections, 1 plaquetopenia, 1 pernicious anemia, 1 corneal ulcer and 1 primary biliary cirrhosis. The majority of the patients received glucocorticoid therapy (12 at a low dose and 16 at a high dose), 7 needed antimalarial treatment and 10 immunosupressive treatment. One patient needed a pulmonary transplant.
Mean ESSDAI was 4.2 (2–9).
DiscussionAbove the glandular manifestations, present in almost all PSS patient, a wide range of extraglandular clinical syndromes may appear in longstanding diseases with a heavy autoimmune background. PSS can affect peripheral and central nervous system, kidney, skin, musculoskeletal and the lungs. Lung disease in the PSS patients can affect their superior and lower respiratory tract, the diaphragm, and the pulmonary vessels. Furthermore, lung lymphoproliferative disease can also be found.
Palm and collegues found a high population-based prevalence of clinical pulmonary involvement (22%) among patients with PSS. They also affirmed that patients with lung involvement had reduced quality of life and increased mortality.9
Patients with extraglandular manifestations frequently can have ILD. Gardiner and collegues wrote that the prevalence of ILD in PSS patients is approximately 8%.11
Recently, Kreider wrote that 10–20% of patients with PSS have lung involvement, and subclinical lung disease is even more frequent and can include evidence of small airway disease and inflammation.2
ILD can be found in various connective tissue diseases. In the PSS, ILD is an extraglandular severe manifestation and it is particularly associated with patients that are positive with anti-Ro and anti-Ro52. In a study done by Ito and collegues6 they found that 61% of 33 patients who had PSS had NSIP in the lung biopsy, 4 had diffuse bronchiolitis, 4 had non-Hodgkin's lymphomas of mucosal associated lymphoid tissue (MALT) and 5 patients had evidence of amyloidosis, but none of these patients had evidence of LIP. LIP was considered one of the most common pulmonary manifestations in PSS, but recent studies have demonstrated a much lower prevalence (0.9–17%)4. The findings from our study show different results from the study done by Ito, although we did not conduct biopsies in all the patients for the diagnosis. We based our diagnosis basically on the HRCT due to findings in the HCRT are used to be well correlated to histopathologic finding.
In patients with active extra-glandular manifestations and in those that are immunologically active it is mandatory to discard the presence of ILD by doing a HRCT and PFTs. The symptoms of ILD are cough, dyspnea, bilateral infiltrates on radiograph and various patters of abnormalities on HRCT scan.8 These exams must be done in the primary stages due to the possible severity of the ILD.
The lung disease can occur before the other more usual manifestations of the syndrome12,13 and also approximately 15% to 20% of patients presenting with chronic ILD can have an occult connective tissue disease.14 In our cohort, in 60% of the patients, the ILD diagnosis was made before the PSS diagnosis.
Although, some factors have been proposed to be associated with a poor outcome in PSS, specifically with a higher risk of lymphoma, high levels of beta2-microglobulin, presence of skin vasculitis, decreased levels of C3, C4 factors, lower level of CD4 lymphocytes and a CD4/CD8 beyond 0.8,15 the levels of b2microglobulin are not associated with the ILD.
The pulmonary manifestation often shows a restrictive pattern and a low DLCO, associated with a high morbidity and a high mortality. In some patients the course of the illness could be benign with a stabilization in the lung disease. Keeping in mind that the PSS with ILD associated to other connective tissue diseases can develop into a pulmonary fibrosis.
It is mandatory a cross and a multi-disciplinary collaboration in the management of any of the connective tissue diseases with lung disease between rheumatologists and pulmonologists. Rheumatologists help to detect an occult connective tissue disease in patients with “idiopathic ILD” by requiring careful attention to the historical clues, the physical examination findings, specific autoantibodies, radiological and histopathological features.16 For pulmonologists it is important to recognize that PSS with ILD can result in severe disease and they can help in the lung disease management. For a multidisciplinary approach pulmonologists need to learn clues to distinguish between idiopathic interstitial pneumonia from connective tissue disease associated with ILD.17
Our study has an important limitation: the information was collected retrospectively.
ConclusionTo summarize, we shall consider that lung involvement can occur in patients with PSS. ILD is one of the extraglandulars manifestations in those patients. In our cohort we observed the following histopathological patterns: NSIP, UIP, OP and LIP (in this order).
Higher rates of mortality and morbidity in PSS are related to the ILD, therefore, patients affected with PSS must be screened to catch a precocious diagnosis doing all the complementary tests needed. It is important to discard a connective tissue disease in patients with ILD. In our cohort the majority of the patients were diagnosed of ILD before being diagnosed of PSS.
Due to the low prevalence of the ILD in PSS, multicentre cohorts are increasingly demanded to give information about the characteristics, the management and treatment not only of the disease, but also of the single organ complications. A multidisciplinary management is needed.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no patient data appear in this article.
Confidentiality of dataThe authors declare that they have followed the protocols of the workplace on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors declare no conflict of interest.


