




We present the case of a 51-year-old woman with a history of several years of non-organ specific Systemic Lupus Erythematosus, who presented a toxic syndrome and adenopathy and cranial nerve affection. We carried out the differential diagnosis. We then described the case resolution and progression.
Presentamos el caso de una mujer de 51 años con antecedentes de lupus eritematoso sistémico no órgano específico de años de evolución, que inicia clínica de síndrome tóxico junto adenopatías y afectación de pares craneales. Desarrollamos el diagnóstico diferencial. Posteriormente, se muestran la resolución del caso y la evolución de la paciente.
We present the case of a 51-year-old female patient with no known drug allergies, who smoked 15 cigarettes per day (cumulative dose: 26Pkg/year) and with a history of myasthenia gravis in 1985, diagnosed following severe muscle fatigue, with resolution following an intervention to resect a thymoma, and systemic lupus erythematosus (SLE) diagnosed in 1990 with malar rash, oral ulcers, nonerosive symmetric, polyarthritis, with involvement of wrists and metacarpophalangeal and proximal interphalangeal joints of both hands for more than 6 weeks, pancytopenia and in which immunological tests highlighted positive antinuclear antibodies (ANA) and anti-DNA, decreased complement (C3 and C4) and RF, negative anti-Ro and La. She was treated with gold salts and glucocorticoids (methylprednisolone 6mg/day) for a year, suspended due to lack of therapeutic response, then treated with methylprednisolone and hydroxychloroquine 4–6mg/day with a good clinical response, maintaining the same treatment until today. She had a history of spontaneous abortion in 1995 and preterm labor with neonatal death within 4 days of birth and identification of positive anticardiolipin antibodies (IgG) in a single determination (being all subsequently negative). She had no previous thromboembolic events. She was also affected by osteoporosis associated with glucocorticoid therapy and menopause. Currently, she was being treated with methylprednisolone 20mg/day, hydroxychloroquine 200mg/day, omeprazole 20mg/day, aspirin 100mg/day, risedronate 35mg/weekly and calcium and vitamin D.
The patient presented constitutional symptoms a month earlier, with loss of 3kg and asthenia. A general lab analysis showed pancytopenia: 2.6 leukocytes×109/l (absolute neutrophils 1.6×109/l, absolute lymphocyte 0.5×109/l), hematocrit (Hct) 27% hemoglobin (Hb) 8.6mg/dl, platelets 105×109/l and elevated acute phase reactants with an erythrocyte sedimentation rate (ESR) of 75mm/h, C-reactive protein (CRP) of 2.75mg/dl. During the month before the onset of symptoms, she visited ophthalmology due to a scotoma in the right visual field and ipsilateral VI cranial nerve palsy with dysesthesias in the territory of the lower jaw and upper right V pair, her physicians opting to hospitalize her for study.
On physical examination, she was hemodynamically stable, afebrile, hydrated, without signs of heart failure, had no significant alterations in cardiopulmonary auscultation and abdominal palpation. The locomotor apparatus showed no signs of inflammatory activity. On neurological examination the presence of cranial nerve VI palsy and no right meningeal signs was highlighted.
The blood count showed leucopenia (0.6×109/l and 1.2 lymphocytes×109/l neutrophils), normocytic anemia (Hb 7.5mg/dl, hematocrit 25% and MCV of 88fl, with 10.7% of hypochromic erythrocytes) and platelets of 86×109/l. The clotting study was normal. Determinations of glucose, renal function, electrolytes, total protein, albumin and muscle enzymes were normal. Liver function tests showed increased total alkaline phosphatase (436U/l) and gamma glutamyl transferase (207U/l). Ferritin and soluble transferrin receptor were elevated, 420ng/ml and 3.21mg/l, respectively. The ESR was 56mm/h and CRP was 3.22mg/dl. The urine was normal, with no evidence of proteinuria in 24h urine. In the study of autoimmunity ANA 1/320, negative anti-DNA, ENA, and RF and anti-CCP. Lupus anticoagulant and anticardiolipin antibodies were negative and complement was within normal ranges. An anomalous band in the gamma protein peak was observed, serum immunofixation showed monoclonal IgG lambda component and urine immunofixation showed no suspicious findings of monoclonality. Tumor markers were requested, chief among them: CA 125: 1530 (normal <40), CA 19.9: 151 (normal <37), beta-2-microglobulin: 9.9mg/l (normal: 0.2 to 2, 3mg/l), CA 153: 58U/ml (normal <40) and HE4 164.7 (normal <150).
A chest X-ray showed a slight impingement of the left diaphragmatic angle without pulmonary parenchymal abnormalities.
For the study of pancytopenia and monoclonal bands we performed a bone marrow aspirate which showed the presence of 3 sets of cellular infiltrate and 6% plasma cells. In a cytospin analysis, lymphocytes with a mature appearance and reactive monocytes and lymphocyte were observed, the result being suggestive of monoclonal gammopathy of undetermined significance. Likewise a bone series was performed in which no osteolytic lesions observed.
For the study of the neurological deficit we requested a computed tomography (CT), which was normal. Magnetic resonance imaging (MRI) revealed a hyperintense central protuberance suggestive of a capillary telangiectasia and a hypointense central area in the spinal cord, from C2 to C5, compatible with syringomyelia. Electromyogram was performed and signs of electrophysiological motor and sensory distal axonopathic polyneuropathy were found.
In order to expand the study of the patient we requested a gynecological ultrasound that showed an endometrial polyp with no other associated abnormalities. The abdomen and pelvis CT revealed the presence of 14cm splenomegaly, with some ill-defined hypodensities, most of 11mm, and multiple bilateral para-aortic and iliac enlarged lymph nodes, most of them measuring 2cm. Chest CT showed mediastinal lymphadenopathy and a nonspecific right apical pulmonary nodule and the presence of emphysema, bronchial thickening and ground-glass opacities that could be related to bronchiolitis.
We performed a diagnostic test.
Differential Diagnosis of the Presenter (Dr. Martinez-Morillo)The current condition of this case comprises essentially a series of hematological and other neurological symptoms. On one hand, the patient has lymphadenopathy, pancytopenia, splenomegaly, elevated β2-microglobulin and a monoclonal band. Likewise, there is also evidence of cranial nerve involvement, polyneuropathy inflammatory joint and spinal fluid. Also noteworthy is the elevation of several tumor markers. Taking multiple adenopathies as a guiding sign, the differential diagnosis should arise between the following diseases (Table 1).
Diseases Accompanied by Adenopathy in Patients With Lupus.
| Infections |
| Mononucleosic syndromes: Epstein–Barr virus, hepatitis B and C virus, human immunodeficiency virus, cytomegalovirus and toxoplasma |
| Tuberculosis |
| Syphilis |
| Leishmania |
| Drugs |
| Systemic diseases |
| Lupus |
| Sarcoidosis |
| Amyloidosis |
| “Benign” lymphoproliferative processes |
| Castleman's syndrome |
| Rosai-Dorfman's disease |
| Kikuchi-Fujimoto's disease |
| Neoplasia |
| POEMS syndrome |
| Carcinoma of unknown origin |
| Non-Hodgkin's lymphoma |
SLE may be complicated by infections1,2 with lymphadenopathy. Infections are the leading cause of death associated to SLE and usually present with fever. They might be confused with a lupus flare or appear simultaneously. Mononucleosis syndromes, such as those caused by Epstein–Barr virus, hepatitis virus B or C, cytomegalovirus, Toxoplasma or the primary infection by the human immunodeficiency virus, present with fever and lymphadenopathy and may present with neurological symptoms. However, in this case specific serology was negative. Disseminated tuberculosis usually occurs in immunosuppressed patients and can affect virtually any organ, including the lymphatic system, bone marrow and nervous system. However, the specific detection of tuberculosis by Elispot® interferon gamma test and cultures for mycobacteria, both of the blood and spinal fluid were negative in this case. Syphilis may also be considered, as it can present as a polyadenopathy syndrome with central nervous system involvement. However, the absence of fever and skin lesions and negative VDRL test result in cerebrospinal fluid rule out this diagnosis. Leishmaniasis can present with lymphadenopathy and pancytopenia, but can be excluded because it affects the nervous system and there was an absence of parasites in the bone marrow aspirate.
Drugs that can cause lymphadenopathy include gold salts, but the chronology of this case does not point to the drug as the cause of the abnormalities.3 Hydroxychloroquine is a very safe drug, but among its side effects are aplastic anemia and peripheral polyneuropathy. However, the central nervous system involvement and lymphadenopathy are not described.4
Lymphadenopathy is part of the clinical spectrum of SLE. They are usually small in size and of cervical, inguinal or axillary location. They are present in up to 25% of patients over the course of the disease and are more frequent at the beginning or during flares.5 However, there are few reported cases in which the main symptom of an outbreak is the polyadenopathy.6 Also noteworthy is the fact that lupus patients have monoclonal bands more often than the general population and that β2-microglobulin is often elevated during flares.7 The outbreak of pancytopenia in lupus is rare; haemolysis, bone marrow infiltration, drug toxicity and hemophagocytic syndrome must be ruled out. Neurological symptoms in this case may also be explained by lupus itself, and as many as 50% of patients present with neurological signs and symptoms throughout the disease. Involvement of cranial nerves and aseptic meningitis are rare, but up to 25% of patients have peripheral polyneuropathy.1 In this case, the outbreak could be ruled out as the cause of lupus manifestations due to normal complement, the negative anti-dsDNA, the response to 20mg of methylprednisolone and the absence of hemolysis and hemophagocytosis in the bone marrow aspirate.
Sarcoidosis can present with hematological and neurological manifestations. Lymphadenopathy is common, as well as bone marrow involvement, although usually mild cytopenias are seen. Chronic meningitis and cranial nerve involvement are common, but peripheral neuropathy is rare. In this case, a bibasal CT pulmonary infiltrate is evident. However, the diagnosis of sarcoidosis should be considered as unlikely by the absence of respiratory symptoms, the severe peripheral pancytopenia8 and the neuropathy. Moreover, in the literature there are very few cases of lupus and sarcoidosis in the same patient. However, definitive diagnosis is histopathological with identification of noncaseating granulomas.
Secondary amyloidosis would be another option. Lupus is an uncommon cause of amyloidosis, and when it occurs, usually the first symptom is proteinuria. It may present with generalized lymphadenopathy and hepatosplenomegaly and possible involvement of the peripheral nervous system by entrapment, but cranial nerves are usually normal.9 Good control of the disease in this case, the low incidence of amyloidosis in lupus, the absence of renal and cranial nerve involvement are against this diagnosis.
Castleman's syndrome is a lymphoproliferative disease characterized by the predominance of CD5 positive B cells in the marginal zone. A multicentric variant may present with lymphadenopathy, hepatosplenomegaly and toxic10 syndrome. There have been reports of coexistence with lupus but these are rare. Only a lymph node biopsy can provide a diagnosis but it can be excluded because cranial nerve involvement and inflammatory cerebrospinal fluid are not described in the literature.
Rosai-Dorfman's disease is a rare condition caused by histiocytic proliferation. It presents with lymphadenopathy, fever, leukocytosis, and monoclonal11 hypergammaglobulinemia. The nervous system involvement is anecdotal, as is its association with lupus.12 Although the probability is very low, to exclude the diagnosis we would require a lymph node biopsy.
Kikuchi-Fujimoto's disease is a histiocytic necrotizing lymphadenitis. The most common site of lymphadenopathy is the cervical region and its evolution is usually benign and self-limited. However, there is widespread agreement that it may cause fever, cytopenias and toxic syndrome, which can be lethal. Its association with lupus is widely described but is very rare.13 Also, though rare, there have been reports of pancytopenia, peripheral neuritis and aseptica14 meningitis, but there is no case where Kikuchi's disease was associated with cranial nerve involvement. Therefore, although this is an unlikely diagnosis, we would require the histopathological examination of a lymph node to definitely dismiss it.
POEMS syndrome (polineuropathy, organomegaly, endocrinopathy, monoclonal proteins and skin changes) or osteosclerotic myeloma lymphadenopathy may occur with a monoclonal lambda type band, as in this case, and peripheral15 polyneuropathy. On the other hand, against the diagnosis of this syndrome we find protein concentrations below 100mg/dl, absence of skin changes or endocrinopathies and absence of sclerotic lesions.
Metastatic carcinoma of unknown origin accounts for 2% of all malignancies and metastases are usually microscopic and obviously from the primary tumor. In this case, the presence of a lung nodule in a CT and a history of smoking may be considered a potential source of pulmonary tumor. The central nervous system involvement could be explained by leptomeningeal infiltration or paraneoplastic syndromes. The patient in this case presented supra and infradiaphragmatic polyadenopathies but did not follow a specific territory that pointed toward a possible primary tumor. Moreover, bone marrow infiltration would be very rare, in order to explain the pancytopenia, and lymphadenopathy, with no evidence of solid organ involvement, so this diagnosis is excluded.2
Lupus is associated with an increased risk of malignancies, especially lymphoma, which may present with lymphadenopathy. In a study by Bernatski et al.16 it was shown that lupus patients had four times the risk of developing non-Hodgkin lymphoma than the general population. NHL presents with lymphadenopathy and hepatosplenomegaly, B-symptoms (fever, weight loss and night sweats), increased LDH and β2-microglobulin, paraneoplastic polyneuropathy, pulmonary infiltration or central nervous system involvement, and a monoclonal gammopathy in the case of mature B cells. Infiltration of the bone marrow that causes cytopenias is also frequent. β2-microglobulin is a relatively sensitive but nonspecific marker of tumor and has prognostic value in lymphoproliferative17 diseases. CEA 125 has also been used to monitor some lymphomas. Pathologic confirmation is required for the diagnosis of lymphoma.
Clinical Diagnosis of the Presenter (Dr. Martinez-Morillo)Integrating data and clinical symptoms of the patient, it seems reasonable to rule out a lymphoproliferative disease as the cause of his present illness.
The diagnosis of non-Hodgkin lymphoma would be plausible due to the presence of lymphadenopathy and splenomegaly, monoclonal bands, malaise, weight loss, elevated LDH, β2-microglobulin, CEA 125 and hepatic infiltration. In addition, the age of the patient, and a history of SLE and thymoma increase the risk of developing this type of tumor.
The severe pancytopenia could be explained by the infiltration of the bone marrow. This involvement is common but patchy, so aspiration may not be enough and in many cases, requires a bone marrow biopsy with immunohistochemical stains for diagnosis. Neurologic manifestations of both the central and peripheral nervous system may be explained by infiltration or paraneoplastic syndromes. Central nervous system involvement may not be visible with unenhanced MRI and lumbar puncture is often not useful to isolate malignant cells. Involvement of cranial nerve V, also known as “numb chin syndrome” (asleep chin syndrome), is important and typically has been associated with occult neoplasia, specifically lymphomas.
Therefore, my definitive diagnosis is of non-Hodgkin lymphoma.
A diagnostic lymph node biopsy would be necessary.18 Needle aspiration may be indicative but rarely diagnostic. If the axillary or inguinal lymphadenopathy detected by CT is of significant size, this would be the first choice. Otherwise, a node biopsy accessing the subcarinal adenopathy may be the best alternative.
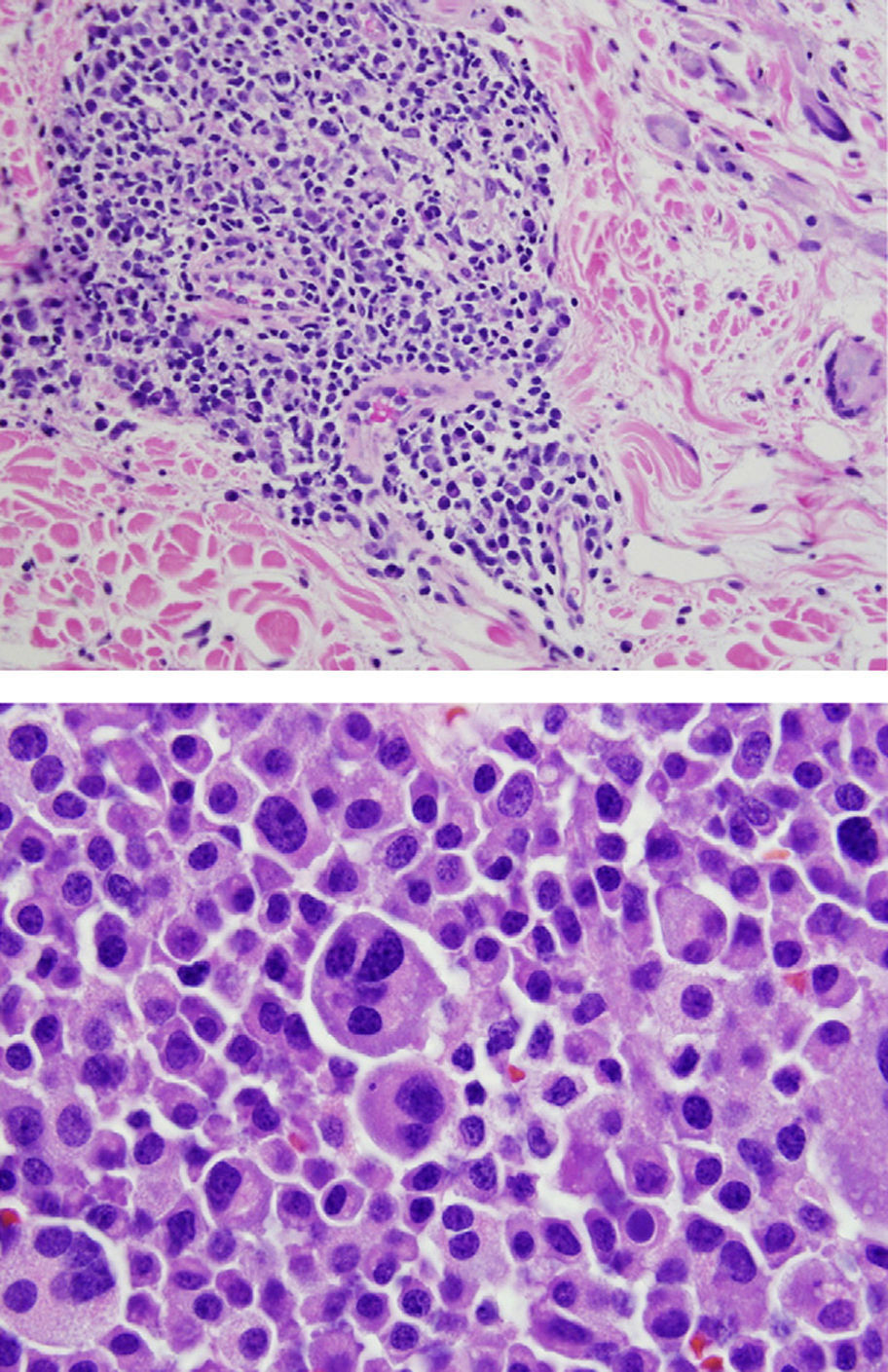
Final Diagnosis of the Presenter (Dr. Gomez Caballero)First, a PET SCAN was performed which showed the presence of multiple laterocervical, mediastinal, abdominal and inguinal hypermetabolic lymph nodes (Fig. 1). But the definite diagnostic test was a lymph node biopsy, where proliferation was observed in lymphoid cells which formed large immunoblastic atypical cells, some with irregular nuclei and abundant cytoplasm with varying degrees of secretory transformation. Accompanying the tumor growth we observed an atypical plasma cell differentiation; these findings support plasmablastic lymphoma (Fig. 2).
Comment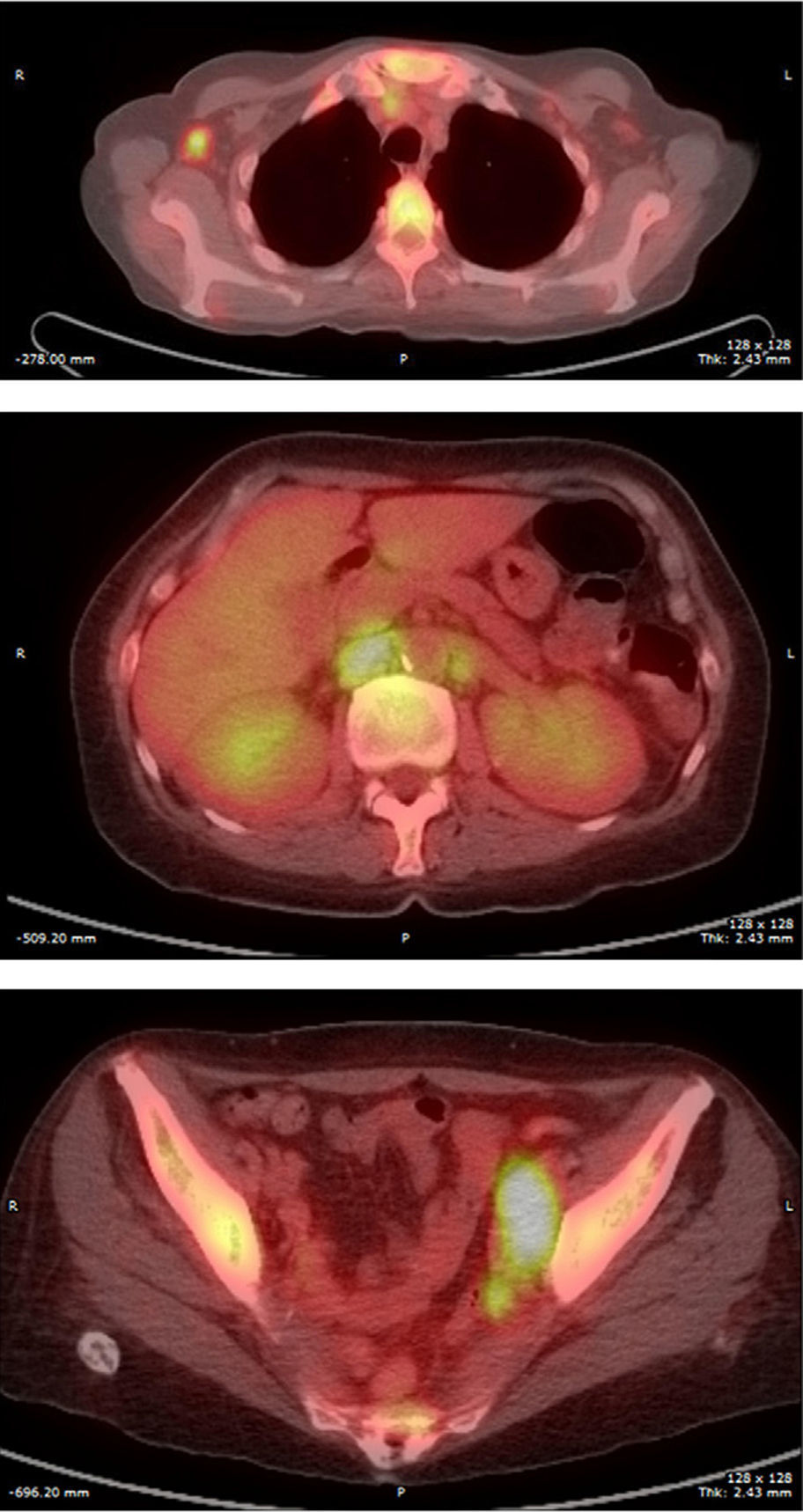
The survival of patients with SLE has changed in recent decades and this has been attributed to an early diagnosis, early treatment and disease control. As a result, the increase in life expectancy is demonstrated by the presence of chronic and late complications, such as neoplasms.
In some types of autoimmune disease, including SLE, it has been shown that hematologic tumors contribute to increased morbidity and mortality.
Recent research shows that the incidence of non-Hodgkin lymphoma (NHL) in patients with SLE is 2–4 times higher than in the general population.
The mechanisms responsible for the association between lymphoma and SLE are still unknown, but it is possible that the altered clearance of apoptotic cells in bone marrow and lymph nodes of patients with SLE influence extended stimulation of reactive B cells, increasing the risk of DNA structural injury and lymphogenesis. Therefore, it has been suggested that there might be a relationship between the inflammatory activity of the disease and increased risk of developing neoplastic disease in these patients.19
Immunosuppressive therapy has also been proposed as a predisposing factor for the development of lymphoma; however, there are cases of patients exposed to these drugs with this complication. Bernatsky et al. analyzed the incidence of cancer in patients with SLE and observed increased risk for the development of hematologic neoplasms in the first year after the diagnosis of SLE, suggesting that the risk of developing this type of cancer would not be totally related to the cumulative dose of imunosuppresive20 treatment. In this sense, the presence of clinical features of patients with SLE including hematologic (hemolytic anemia), immunological (antibody positivity anti-Ro/La), clinical manifestations (symptoms suggestive of secondary Sjögren's syndrome), as well as pulmonary infiltrates and/or pneumonia, all have been associated with an increased risk of developing NHL.21
Clinically and histologically more aggressive forms, particularly Diffuse large B cell lymphoma, constitute approximately 30% of patients diagnosed with NHL, this being the most representative subtype (over 50%) in patients diagnosed with SLE and lymphoproliferative disease. There are also other subtypes of NHL in patients with lupus; these subtypes include the diagnosis in our patient, “plasmablastic lymphoma”, which is considered a rare histologic variant of diffuse large cell type B lymphoma. This type of lymphoma usually occurs in immunocompromised patients, has an aggressive clinical course and poor prognosis, with a median overall survival of 15 months and a mortality rate between 50% and 60%. Although it can be seen in other locations, it usually occurs in the oral cavity and/or jaw, with rare lymph node involvement, which confers an additional peculiarity of our case as it presented with node involvement.22
The treatment of lymphoproliferative diseases in SLE patients differs from patients without the disease.
Identification of NHL in SLE patients may be difficult because the two entities share similar clinical characteristics, such as lymphadenopathy, fever, weight loss, hepato/splenomegaly and/or cytopenias, among others. Only close monitoring of patients with SLE may help detect early clinical and laboratory data indicative of neoplastic disease.
The clinical course of our patient after diagnosis was unfavorable, as expected by the histological type and staging of lymphoma. During her hospitalization, she presented persistent fever and signs of respiratory failure, and was transferred to the intensive care unit under the tentative diagnosis of septic shock, probably of respiratory origin. She subsequently required intubation, mechanical ventilation support, with broad-spectrum antibiotics and vasoactive drugs. In the absence of growth of microorganisms in blood cultures and bronchoalveolar lavage, it was decided to initiate CHOP chemotherapy. However, she did not adequately respond to treatment, showing sustained severe pancytopenia, multiple organ failure and death.
Ethical disclosuresProtection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of Data. The authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consent. The authors declare that no patient data appears in this article.
Conflict of InterestThe authors declare no conflict of interest.
Please cite this article as: Gómez Caballero ME, Martínez-Morillo M. Mujer con lupus eritematoso sistémico y poliadenopatías. Reumatol Clin. 2012. http://dx.doi.org/10.1016/j.reuma.2012.06.012.


