


To assess the prevalence of systemic and organ-specific autoimmunity among individuals with human inborn errors of immunity (IEI).
MethodsRetrospective study. We recorded demographic variables, type of immunodeficiency, and systemic and organ specific autoimmunity.
ResultsWe included 48 patients (54.1% men) with mean age of 32.1 years. The most common IEIs included combined immunodeficiency with syndromic features (31.2%) and predominantly antibody deficiency (20.1%). We observed autoimmunity in 15 patients (31.2%): 12 organ-specific autoimmunity and 5 systemic autoimmunity, not mutually exclusive groups. Organ-specific autoimmunity preceded the onset of IEI in 5 patients, was concurrent in one patient, and developed after the diagnosis of IEI in 6 cases. From the systemic autoimmunity group, we observed polyarteritis nodosa (n = 2), antiphospholipid syndrome (APS) (n = 2), and overlap of limited systemic sclerosis/APS/Sjögren's syndrome (n = 1), and in all cases, this occurred after the IEI diagnosis.
ConclusionOur findings confirm the coexistence of autoimmunity and IEI. This overlap may be attributed to B and T cell disorders, as well as potential alterations in the microbiota in these patients.
Evaluar la prevalencia de autoinmunidad sistémica e inmunidad órgano-específica en pacientes con errores innatos de la inmunidad (EII) en un centro de tercer nivel de atención.
MétodosEstudio retrospectivo. Se registraron variables demográficas, tipo de inmunodeficiencia, y autoinmunidad órgano-específica y sistémica.
ResultadosSe incluyeron 48 pacientes (54.1% hombres) con edad promedio de 32.1 años. Las EIIs más frecuente fueron inmunodeficiencia combinada con características sindrómicas (31.2%) y deficiencia predominantemente de anticuerpos (20.1%). Documentamos autoinmunidad en 15 pacientes (31.2%): 12 órgano-específica y 5 sistémica, grupos no mutuamente excluyentes. La autoinmunidad órgano-específica precedió en 5 pacientes a la EII, en 1 fue concomitante y en 6 posterior. Del grupo de autoinmunidad sistémica observamos poliarteritis nodosa (n = 2), síndrome antifosfolípido (SAF) (n = 2) y sobreposición esclerosis sistémica limitada/SAF/Síndrome de Sjögren (n = 1), y en todos fue posterior a la EII.
ConclusiónNuestros hallazgos apoyan la coexistencia de autoinmunidad en EII. Esta coexistencia podría deberse a una alteración inmunológica en células B y T, así como al posible papel de una microbiota alterada en estos pacientes.
Inborn errors of immunity (IEI) encompass inherited disorders of the function and regulation of the immune system characterised by increased susceptibility to infections, autoinflammation, lymphoproliferation, granuloma formation, atopy and malignancy.1,2 While these patients have been reported to be prone to autoimmunity,3 this has been focused on a specific IEI.
With respect to organ-specific autoimmunity, in a cohort of 562 patients with IEI, 6% had some autoimmune cytopenia.2 In contrast, there have been only one-off reports of systemic autoimmunity.
This study has assessed the prevalence of systemic and organ-specific autoimmunity in a cohort of adult patients with IEI.
Material and methodsA retrospective case series of subjects with IEI following the classification of the Committee of Experts of the International Union of Immunological Societies1 who attended a tertiary public healthcare centre between January 2006 and March 2023 and who had a complete clinical record available for review. Demographic variables, type of immunodeficiency, duration, organ-specific autoimmunity (thyroid, hepatic, renal, dermatological, endocrinological, neurological, and haematological), systemic autoimmunity, and time of debut were all recorded. Descriptive statistics were used based on the distribution of the variables. The study was approved by the Institutional Ethics Committee.
Forty-eight patients (26 males, 54.1%) with IEI, mean current age of 32.1 ± 13 years, and mean time of evolution of 16.9 ± 9.1 years were identified and collected. The most frequent IEIs were combined immunodeficiency with syndromic features (31.2%) and predominantly antibody deficiency (20.1%) (Fig. 1).

We documented systemic and/ or organ-specific autoimmunity in 15 patients (31.2%), 12 of which were organ-specific and five were systemic; these groups were not mutually exclusive.
Organ-specific autoimmunity preceded the IEI in five cases (median 7 years, range 1–19), was concomitant in one, and posterior in six (median 4 years, range 1–17). We observed immune thrombocytopenia (n = 3), autoimmune thyroid disease (n = 5), alopecia areata (n = 1), vitiligo (n = 2), morphea (n = 1), autoimmune hepatitis (n = 1), ulcerative colitis (n = 1), Evans syndrome (n = 2), pernicious anaemia (n = 1), and myasthenia gravis (n = 1).
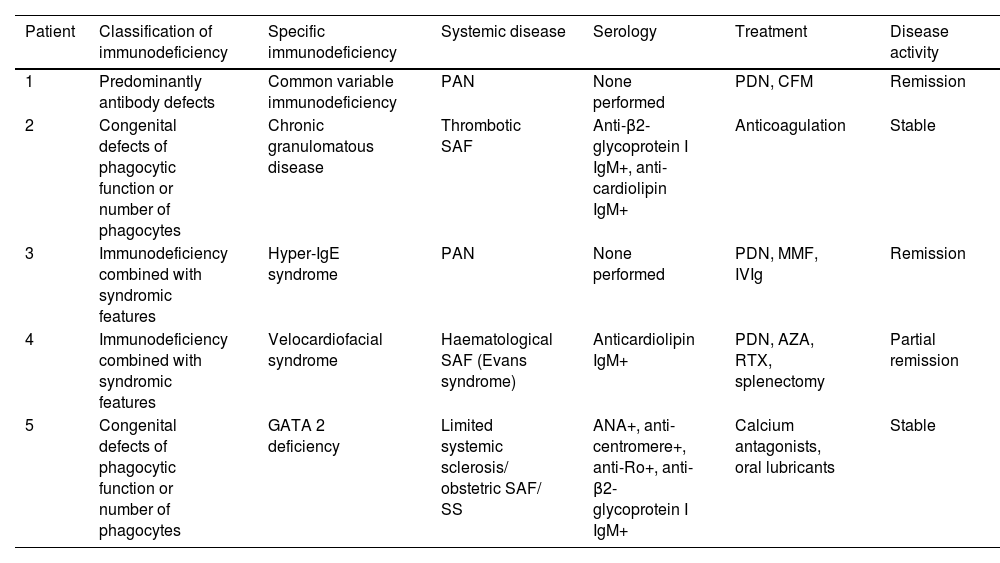
Of the participants with systemic autoimmunity, we detected polyarteritis nodosa (PAN) (n = 2), antiphospholipid syndrome (APS) (n = 2), and limited systemic sclerosis/ SAF/ Sjögren's syndrome (SS) overlap (n = 1); in all of them, it was following the IEI (median 8 years, range 1–33 years). Tabla 1 displays the patient characteristics.
Patients with inborn errors of immunity and systemic autoimmune diseases.
| Patient | Classification of immunodeficiency | Specific immunodeficiency | Systemic disease | Serology | Treatment | Disease activity |
|---|---|---|---|---|---|---|
| 1 | Predominantly antibody defects | Common variable immunodeficiency | PAN | None performed | PDN, CFM | Remission |
| 2 | Congenital defects of phagocytic function or number of phagocytes | Chronic granulomatous disease | Thrombotic SAF | Anti-β2- glycoprotein I IgM+, anti-cardiolipin IgM+ | Anticoagulation | Stable |
| 3 | Immunodeficiency combined with syndromic features | Hyper-IgE syndrome | PAN | None performed | PDN, MMF, IVIg | Remission |
| 4 | Immunodeficiency combined with syndromic features | Velocardiofacial syndrome | Haematological SAF (Evans syndrome) | Anticardiolipin IgM+ | PDN, AZA, RTX, splenectomy | Partial remission |
| 5 | Congenital defects of phagocytic function or number of phagocytes | GATA 2 deficiency | Limited systemic sclerosis/ obstetric SAF/ SS | ANA+, anti-centromere+, anti-Ro+, anti- β2-glycoprotein I IgM+ | Calcium antagonists, oral lubricants | Stable |
ANA: antinuclear antibodies; AZA: azathioprine; CFM: cyclophosphamide; MMF: mycophenolate mofetil; PAN: polyarteritis nodosa; PDN: prednisone; RTX: rituximab; SAF: antiphospholipid syndrome; SS: Sjögren’s syndrome.
The CEREDIH registry (n = 2183) of IEI has documented autoimmune disorders in 26.2% of the patients, with the risk of autoimmune disease being 10 times greater than in the general population, and higher in common variable immunodeficiency (CVID) and T4 cell deficiency. The USIDNET cohort of individuals with CVID (n = 870) reported autoimmunity in 5.9%, most frequently affecting females and non-whites, and not associated with CD19+ B-lymphocyte count, CD4/ CD8 ratio or serum immunoglobulins. Most had inflammatory arthritis (n = 18) and SS (n = 11), followed by lupus and vasculitis.5 Other studies have indicated a prevalence rate of rheumatoid arthritis and juvenile idiopathic arthritis in CVID of 1–5%,6–8 and there are isolated cases with inflammatory myopathies and relapsing polychondritis.9 In primary antibody deficiencies (CVID and other conditions), up to 16% of patients present with spondyloarthritis-like arthritis or enteropathic arthritis,7 whereas in autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy, SS is most common.10 As for organ-specific autoimmunity in CVID, gastrointestinal and haematological autoimmunity (haemolytic anaemia, thrombocytopenia, neutropenia, and pernicious anaemia) are the predominant manifestations.6,10
In our study involving a variety of IEIs, we documented 31.2% of systemic and/ or organ-specific autoimmunity. The time of onset was variable in the organ-specific autoimmunity group but was after the diagnosis of IEI in all cases of systemic autoimmunity. Our case series exhibited PFS and PAN more frequently, in contrast to the literature.
ConclusionOur findings corroborate the coexistence of autoimmunity in individuals with IEI. This co-existence may be due to an immunological alteration of B and T cells, or to an alteration of the microbiota.
FundingThis research has received no specific grants from public-sector agencies, the commercial sector, or from non-profit organisations.
Conflict of interestsThe authors have no conflict of interests to declare.


