


Evaluar la prevalencia de autoinmunidad sistémica e inmunidad órgano-específica en pacientes con errores innatos de la inmunidad (EII) en un centro de tercer nivel de atención.
MétodosEstudio retrospectivo. Se registraron variables demográficas, tipo de inmunodeficiencia, y autoinmunidad órgano-específica y sistémica.
ResultadosSe incluyeron 48 pacientes (54,1% hombres) con edad promedio de 32,1 años. Los EII más frecuentes fueron inmunodeficiencia combinada con características sindrómicas (31,2%) y deficiencia predominantemente de anticuerpos (20,1%). Documentamos autoinmunidad en 15 pacientes (31,2%): 12 órgano-específica y 5 sistémica, grupos no mutuamente excluyentes. La autoinmunidad órgano-específica precedió en 5 pacientes a los EII, en uno fue concomitante y en 6 posterior. Del grupo de autoinmunidad sistémica observamos poliarteritis nodosa (n=2), síndrome antifosfolípido (SAF) (n=2) y sobreposición esclerosis sistémica limitada/SAF/síndrome de Sjögren (n=1), y en todos fue posterior a los EII.
ConclusiónNuestros hallazgos apoyan la coexistencia de autoinmunidad en EII. Esta coexistencia podría deberse a una alteración inmunológica en células B y T, así como al posible papel de una microbiota alterada en estos pacientes.
To assess the prevalence of systemic and organ-specific autoimmunity among individuals with human inborn errors of immunity (IEI).
MethodsRetrospective study. We recorded demographic variables, type of immunodeficiency, and systemic and organ specific autoimmunity.
ResultsWe included 48 patients (54.1% men) with mean age of 32.1 years. The most common IEIs included combined immunodeficiency with syndromic features (31.2%) and predominantly antibody deficiency (20.1%). We observed autoimmunity in 15 patients (31.2%): 12 organ-specific autoimmunity and 5 systemic autoimmunity, not mutually exclusive groups. Organ-specific autoimmunity preceded the onset of IEI in 5 patients, was concurrent in one patient, and developed after the diagnosis of IEI in 6 cases. From the systemic autoimmunity group, we observed polyarteritis nodosa (n=2), antiphospholipid syndrome (APS) (n=2), and overlap of limited systemic sclerosis/APS/Sjögren's syndrome (n=1), and in all cases, this occurred after the IEI diagnosis.
ConclusionOur findings confirm the coexistence of autoimmunity and IEI. This overlap may be attributed to B and T cell disorders, as well as potential alterations in the microbiota in these patients.
Los errores innatos de la inmunidad (EII) comprenden trastornos hereditarios de la función y regulación inmunitaria caracterizados por mayor susceptibilidad a infecciones, autoinflamación, linfoproliferación, formación de granulomas, atopia y malignidad1,2. Si bien se ha descrito predisposición a la autoinmunidad en estos pacientes3, ésta se ha centrado en algún EII en específico. En cuanto a la autoinmunidad órgano-específica, en una cohorte de 562 pacientes con EII, el 6% presentó alguna citopenia autoinmune2. Por otra parte, los reportes de autoinmunidad sistémica son aislados.
Este estudio evaluó la prevalencia de autoinmunidad sistémica y órgano-específica en una cohorte de pacientes adultos con EII.
Material y métodosSerie de casos retrospectiva de pacientes con EII según la clasificación del Comité de Expertos de la Unión Internacional de Sociedades de Inmunología1 que acudieron a un centro de tercer nivel de atención pública en el período enero 2006-marzo 2023 y que contaban con expediente clínico completo para su revisión. Se registraron variables demográficas, tipo de inmunodeficiencia, años de evolución, autoinmunidad órgano-específica (tiroidea, hepática, renal, dermatológica, endocrinológica, neurológica y hematológica), autoinmunidad sistémica, y momento de presentación. Se utilizó estadística descriptiva de acuerdo a la distribución de las variables. El estudio fue aprobado por el Comité de Ética institucional.
Se identificaron e incluyeron 48 pacientes (26 hombres, 54,1%) con EII, edad actual de 32,1±13 años, tiempo de evolución de 16,9±9,1 años. Los EII más frecuentes fueron la inmunodeficiencia combinada con características sindrómicas (31,2%) y la deficiencia predominantemente de anticuerpos (20,1%) (fig. 1).

Documentamos autoinmunidad sistémica y/u órgano-específica en 15 pacientes (31,2%), de los cuales 12 fueron órgano-específica y 5 sistémica, grupos no mutuamente excluyentes.
La autoinmunidad órgano-específica precedió en 5 pacientes a los EII (mediana 7 años, rango 1-19), en uno fue concomitante y en 6 posterior (mediana de 4 años, rango 1-17). Observamos trombocitopenia inmune (n=3), enfermedad tiroidea autoinmune (n=5), alopecia areata (n=1), vitíligo (n=2), morfea (n=1), hepatitis autoinmune (n=1), colitis ulcerativa (n=1), síndrome de Evans (n=2), anemia perniciosa (n=1) y miastenia gravis (n=1).
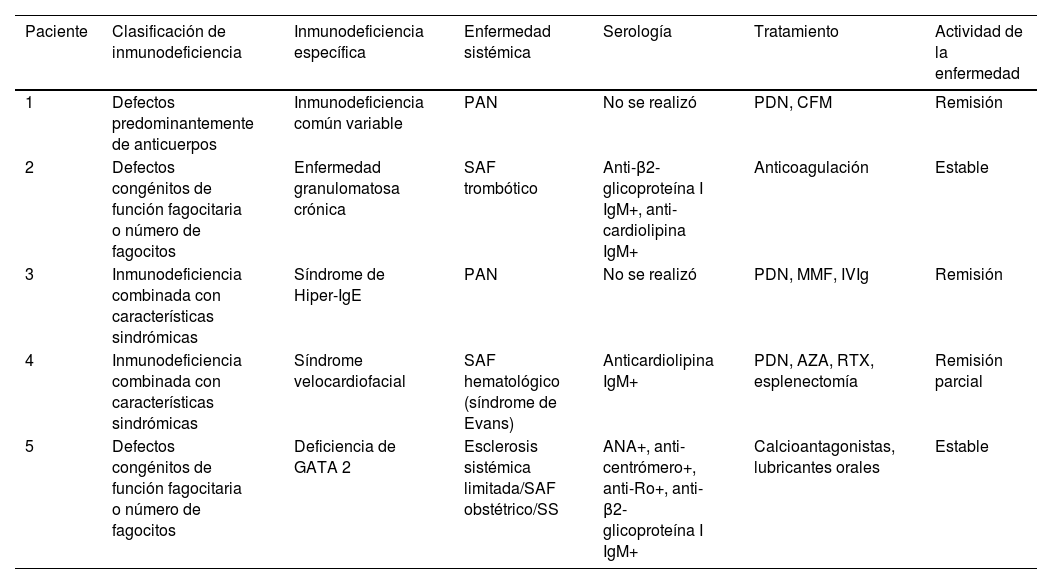
De los pacientes con autoinmunidad sistémica observamos poliarteritis nodosa (PAN) (n=2), síndrome antifosfolípido (SAF) (n=2) y sobreposición esclerosis sistémica limitada/SAF/síndrome de Sjögren (SS) (n=1), y en todos fue posterior a los EII (mediana 8 años, rango 1-33 años). La tabla 1 muestra las características de los pacientes.
Pacientes con errores innatos de la inmunidad y enfermedades autoinmunes sistémicas
| Paciente | Clasificación de inmunodeficiencia | Inmunodeficiencia específica | Enfermedad sistémica | Serología | Tratamiento | Actividad de la enfermedad |
|---|---|---|---|---|---|---|
| 1 | Defectos predominantemente de anticuerpos | Inmunodeficiencia común variable | PAN | No se realizó | PDN, CFM | Remisión |
| 2 | Defectos congénitos de función fagocitaria o número de fagocitos | Enfermedad granulomatosa crónica | SAF trombótico | Anti-β2-glicoproteína I IgM+, anti-cardiolipina IgM+ | Anticoagulación | Estable |
| 3 | Inmunodeficiencia combinada con características sindrómicas | Síndrome de Hiper-IgE | PAN | No se realizó | PDN, MMF, IVIg | Remisión |
| 4 | Inmunodeficiencia combinada con características sindrómicas | Síndrome velocardiofacial | SAF hematológico (síndrome de Evans) | Anticardiolipina IgM+ | PDN, AZA, RTX, esplenectomía | Remisión parcial |
| 5 | Defectos congénitos de función fagocitaria o número de fagocitos | Deficiencia de GATA 2 | Esclerosis sistémica limitada/SAF obstétrico/SS | ANA+, anti-centrómero+, anti-Ro+, anti- β2-glicoproteína I IgM+ | Calcioantagonistas, lubricantes orales | Estable |
ANA: anticuerpos antinucleares; AZA: azatioprina; CFM: ciclofosfamida; MMF: micofenolato de mofetilo; PAN: poliarteritis nodosa; PDN: prednisona; RTX: rituximab; SAF: síndrome antifosfolípido; SS: síndrome de Sjögren.
El registro CEREDIH (n=2183) de EII reportó alteraciones autoinmunes en el 26,2% de los pacientes, siendo el riesgo de enfermedad autoinmune 10 veces más que la población general, y mayor en inmunodeficiencia común variable (IDCV) y deficiencia de células T4. La cohorte USIDNET de pacientes con IDCV (n=870) reportó autoinmunidad en el 5,9%, afectando más frecuentemente a mujeres y no blancos, y no se asoció a la cuenta de linfocitos B CD19+, razón CD4/CD8 o inmunoglobulinas séricas. La mayoría presentó artritis inflamatoria (n=18) y SS (n=11), seguidos por lupus y vasculitis5. En otros estudios, la prevalencia de artritis reumatoide y artritis idiopática juvenil en IDCV es del 1-5%6–8, y existen casos aislados con miopatías inflamatorias y policondritis recidivante9. En deficiencias primarias de anticuerpos (IDCV y otras entidades), hasta el 16% de pacientes presentan artritis similar a espondiloartritis o artritis enteropática7, mientras que en la poliendocrinopatía autoinmune con candidiasis y distrofia ectodérmica lo más frecuente es el SS10. Referente a autoinmunidad órgano-específica en IDCV, predomina la gastrointestinal y hematológica (anemia hemolítica, trombocitopenia, neutropenia y anemia perniciosa)6,10.
En nuestro estudio que incluyó diversos EII, documentamos un 31,2% de autoinmunidad sistémica y/u órgano-específica. El momento de aparición fue variable en el grupo de autoinmunidad órgano-específica, pero fue posterior al diagnóstico de EII en todos los casos de autoinmunidad sistémica. Nuestros pacientes presentaron con mayor frecuencia SAF y PAN, en contraste con la literatura.
ConclusiónNuestros hallazgos respaldan la coexistencia de la autoinmunidad en pacientes con EII. Esta coexistencia podría deberse a una alteración inmunológica en las células B y T, o a una alteración de la microbiota.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores no tienen ningún conflicto que declarar.


