


Mutations in the α-galactosidase A (GLA) gene result in Fabry disease (FD), a rare metabolic condition. FD patients present with heterogeneous clinical manifestations, which may overlap with systemic diseases including familial Mediterranean fever (FMF). The aim of this study was to determine the frequency of FD in patients with mild and severe FMF and to prevent misdiagnosis by increasing clinicians’ awareness.
MethodsBased on Tel-Hashomer criteria, the study included a total of 91 FMF patients. Patients were divided into two groups according to the number of recurrent clinical episodes or failure to respond to maximum therapy: those with mild and severe forms of the disease. GLA gene mutations and α-GLA enzyme activity were assessed. Records of MEFV mutations, therapies and demographic characteristics were kept.
ResultsFD testing was performed on a cohort of 91 FMF patients, 54.9% had mild FMF, 45.1% had severe FMF, and only one patient in the mild FMF subgroup tested positive for FD. The patient was a 39-year-old woman with a history of recurrent abdominal pain, distal limb pain and fever. She had low GLA enzyme activity and a heterozygous GLA gene mutation.
ConclusionsOur findings suggest that FD should be considered in the differential diagnosis of FMF, especially in individuals with unusual symptoms.
Las mutaciones en el gen de la α-galactosidasaA (GLA) dan lugar a la enfermedad de Fabry (EF), un trastorno metabólico poco frecuente. Los pacientes con EF presentan manifestaciones clínicas heterogéneas que pueden solaparse con enfermedades sistémicas como la fiebre mediterránea familiar (FMF). El objetivo de este estudio era determinar la frecuencia de la enfermedad de EF en pacientes con FMF leve y grave y evitar diagnósticos erróneos mediante una mayor concienciación de los médicos.
MétodosBasándose en los criterios de Tel-Hashomer, el estudio incluyó un total de 91 pacientes con FMF. Los pacientes se dividieron en dos grupos según el número de episodios clínicos recurrentes o la falta de respuesta al tratamiento máximo: los que presentaban formas leves (grupo1) y graves (grupo2) de la enfermedad. Se evaluaron las mutaciones del gen GLA y la actividad de la enzima α-GLA. Se mantuvieron registros de las mutaciones del MEFV, las terapias y las características demográficas.
ResultadosSe realizaron pruebas de EF en una cohorte de 91 pacientes con FMF. El 54,9% presentaban FMF leve, el 45,1% FMF grave, y solo un paciente del subgrupo de FMF leve dio positivo en las pruebas de EF. Se trataba de una mujer de 39años con antecedentes de dolor abdominal recurrente, dolor en las extremidades distales y fiebre. Tenía una actividad enzimática GLA baja (0,1nmol/h/ml [normal: >1,37nmol/h/ml) y una mutación heterocigota del gen GLA. Sin embargo, la prueba de la mutación MEFV fue negativa. En su historial detallado de enfermedad familiar refería acroparestesia leve, hipohidrosis y acufenos.
ConclusionesNuestros hallazgos sugieren que la EF debe considerarse en el diagnóstico diferencial de la FMF, especialmente en individuos con síntomas inusuales.
Fabry disease (FD) is a rare X-linked lysosomal storage disorder resulting from deficient activity of the α-galactosidase A (GLA) enzyme encoded by the GLA gene located at Xq22.1. This insufficiency leads to the progressive accumulation of globotriaosylceramide (GL-3 or Gb3) in various cell types, including capillary endothelial, renal, cardiac, and neural cells.1 Tissue damage typically initiates in childhood, yet most patients remain clinically asymptomatic during their early years, distinguishing it from many other lysosomal storage disorders that manifest early in life.2 Generally, men experience more severe symptoms due to the X-linked recessive inheritance pattern, resulting in low or absent enzyme activity. Heterozygous women may exhibit a variable disease course, ranging from an asymptomatic state to a severe phenotype resembling that seen in men.3 LysoGb3, a degradation product of the accumulated Gb3 in lysosomes, is believed to possess cytotoxic, proinflammatory, and profibrotic properties. Several symptoms associated with this metabolite, such as fever, gastrointestinal symptoms, proteinuria, renal failure, and left ventricular hypertrophy, may manifest throughout the course of the disease.2,4
Recurrent acute episodes of fever with serositis are the hallmark of familial Mediterranean fever (FMF), an autoinflammatory disease. It is an autosomal recessive disorder caused by mutations in the MEFV pyrin coding gene located on chromosome 16p13.3.5 The clinical presentation of FMF can vary among patients, with some experiencing mild symptoms and others having frequent attacks, which can lead to complications like amyloidosis.
The common pathophysiological pathway between FD and FMF is primarily their involvement in inflammatory processes, although the underlying causes of these processes differ significantly. This common feature, in particular the role of chronic inflammation, can sometimes lead to overlapping clinical presentations, especially in the context of vascular and systemic involvement. Both diseases are associated with chronic inflammation. In FD, Gb3 accumulation can trigger inflammatory processes in tissues, especially in the vascular endothelium, and contribute to vascular damage and organ dysfunction.6 In FMF, inflammation is the central feature, driven by excessive IL-1 production resulting from pyrin dysregulation. Although the primary mechanisms are different (lysosomal storage in FD and dysregulated inflammation in FMF), there are similarities in the components of inflammation.7
In the TURKFAB study, Turkmen et al.8 found the prevalence of FD to be 0.95% in patients with chronic kidney disease of unknown etiology. It was highlighted that FMF is a common misdiagnosis, resulting in a five-year delay in final diagnosis. Throughout the world, the FMF disease is most frequently seen in Turkey with a prevalence varying between 1:150 and 1:1000.9 Some symptoms of FD, such as painful abdominal attacks and fever episodes, might be mistaken for FMF in the absence of a severe clinical picture. This could result in an underestimating of FD because FMF would have been misdiagnosed.10 This difficulty in correctly differentiating between these diseases supports the idea that all patients with a suspected diagnosis of FMF should have their GLA and MEFV genes analysed, in addition to a careful family history, in case their diagnosis is not genetically justified.11,12
Therefore, we aimed to determine the spectrum of mutations present in the MEFV and GLA genes to identify cases of possible misdiagnosis of FMF and/or missed diagnosis of FD. The objective is to enhance awareness of this diagnostic challenge and offer valuable insights for clinicians and researchers.
Patients and methodsPatients included in this study were drawn from our outpatient clinic. Eligible participants were adult individuals diagnosed with FMF according to the Tel-Hashomer criteria.13 Selection criteria also required patients to possess complete MEFV gene information and a comprehensive clinical and family history.
Participants were administered a targeted questionnaire designed to capture information related to both FMF and FD such as pain, skin-related issues, gastrointestinal symptoms, cardiovascular concerns, renal involvement, ocular symptoms, and neurological manifestations. Questions included inquiries about specific symptoms, such as burning pain, skin lesions, reduced sweating, nausea, hypertension, proteinuria, and others.
Based on the collected data, patients were categorized into two groups: those with mild and severe forms of the disease. The severe group encompassed individuals experiencing recurrent clinical attacks (averaging one or more attacks per month over a 3-month period) or demonstrating persistent laboratory inflammation between attacks, even in the presence of the maximum tolerated colchicine dosage.14 Additionally, patients with significant proteinuria, documented amyloidosis, renal failure, or those receiving biological agents were classified within the severe subset.
To assess GLA enzyme activity, a dried blood filter paper test was employed following the protocol described by Chamoles et al.15 Blood drops were incubated on dry filter paper, and enzyme analysis was conducted fluorometrically using a substrate derived from 4-methylumbelliferone. Blood spots were allowed to dry for a minimum of 4h at room temperature and were stored in sealed plastic bags at 4°C for up to one week in a designated storage area. A GLA enzyme level of less than 1.37nmol/h/mL was considered to have low GLA enzyme activity.16
For the determination of FD, patients were screened for mutation analysis of the GLA gene. Ethylenediaminetetraacetic acid (EDTA) tubes were used to collect peripheral blood samples for GLA gene processing. The exons of the GLA gene were all sequenced using the Sanger sequencing technique.
Compliance with ethical standardsThe study received the local ethics committee's blessing (Decision No: 2020/9), and all participants provided signed informed permission. All methods used in studies involving human subjects adhered to the ethical requirements of the institutional and/or national research committee, the Helsinki Declaration of 1964 and its subsequent amendments, or comparable ethical standards.
Statistical analysisWe performed statistical analyses using SPSS 18.0 for Windows (IBM Corp., Armonk, NY, USA). Normality was assessed using the Kolmogorov–Smirnov test. Continuous data were reported as medians with corresponding minimum–maximum values, while categorical data were presented as numbers and percentages, as appropriate. Mann–Whitney U tests were employed for group comparisons with continuous data, and for categorical variables, we utilized Fisher's exact test or the chi-square test, as appropriate. A significance level of p<0.05 was applied to all statistical tests.
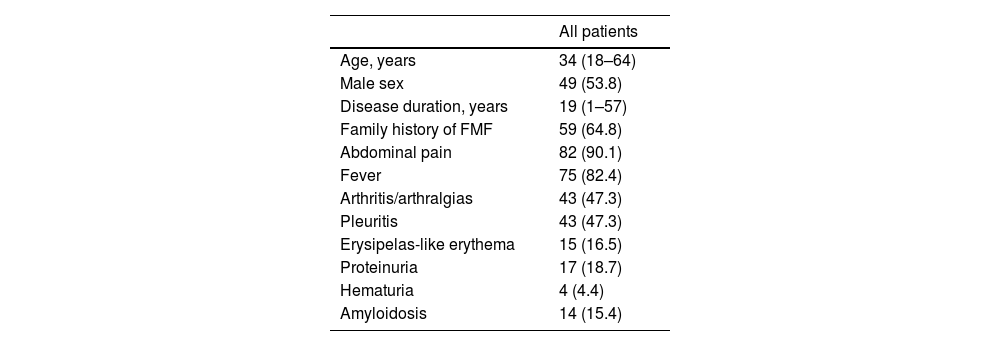
ResultsIn our cohort of 91 patients, the median age was 34 years (range: 18–64), with 53.8% being male. The median disease duration was 19 years (range: 1–57). Table 1 provides an overview of the research group's demographic and clinical features.
Demographic and clinical characteristics of the study group.
| All patients | |
|---|---|
| Age, years | 34 (18–64) |
| Male sex | 49 (53.8) |
| Disease duration, years | 19 (1–57) |
| Family history of FMF | 59 (64.8) |
| Abdominal pain | 82 (90.1) |
| Fever | 75 (82.4) |
| Arthritis/arthralgias | 43 (47.3) |
| Pleuritis | 43 (47.3) |
| Erysipelas-like erythema | 15 (16.5) |
| Proteinuria | 17 (18.7) |
| Hematuria | 4 (4.4) |
| Amyloidosis | 14 (15.4) |
Continuous data are expressed as medians accompanied by the corresponding minimum and maximum values, while categorical data are presented as counts (n) along with the respective percentages in parentheses.
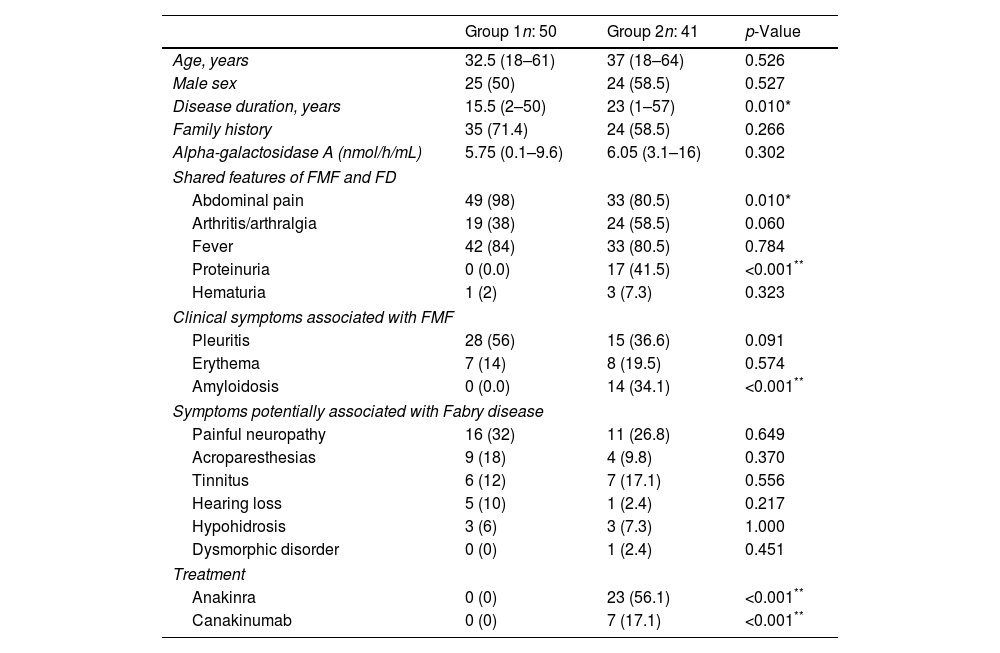
Upon stratifying patients into mild (Group 1, n=50) and severe (Group 2, n=41) FMF categories, both groups exhibited similar sex ratios, ages, and family histories. However, the mild-onset group demonstrated a significantly shorter median disease duration compared to the severe group (15.5 years vs. 23 years, p=0.010). Abdominal pain was more prevalent in the mild-onset group (98% vs. 80.5%, p=0.010), while proteinuria was significantly higher in the severe subtype (41.5% vs. 0.0%, p<0.001). Clinical symptoms associated with FMF, such as pleuritis and erythema, exhibited a trend and no significant difference, respectively. However, amyloidosis was significantly more prevalent in the severe-onset group (34.1% vs. 0%, p<0.001). The median GLA enzyme activity showed no statistically significant difference between mild and severe FMF patients, with values of 5.75nmol/h/mL (range: 0.1–9.6nmol/h/mL) and 6.05nmol/h/mL (range: 3.1–16nmol/h/mL), respectively. Among severe cases, 30 patients were using biological treatments, and 36.7% of these individuals were diagnosed with amyloidosis. Symptoms potentially associated with FD did not significantly differ between the groups (Table 2).
Demographic and clinical findings of patients stratified by disease severity.
| Group 1n: 50 | Group 2n: 41 | p-Value | |
|---|---|---|---|
| Age, years | 32.5 (18–61) | 37 (18–64) | 0.526 |
| Male sex | 25 (50) | 24 (58.5) | 0.527 |
| Disease duration, years | 15.5 (2–50) | 23 (1–57) | 0.010* |
| Family history | 35 (71.4) | 24 (58.5) | 0.266 |
| Alpha-galactosidase A (nmol/h/mL) | 5.75 (0.1–9.6) | 6.05 (3.1–16) | 0.302 |
| Shared features of FMF and FD | |||
| Abdominal pain | 49 (98) | 33 (80.5) | 0.010* |
| Arthritis/arthralgia | 19 (38) | 24 (58.5) | 0.060 |
| Fever | 42 (84) | 33 (80.5) | 0.784 |
| Proteinuria | 0 (0.0) | 17 (41.5) | <0.001** |
| Hematuria | 1 (2) | 3 (7.3) | 0.323 |
| Clinical symptoms associated with FMF | |||
| Pleuritis | 28 (56) | 15 (36.6) | 0.091 |
| Erythema | 7 (14) | 8 (19.5) | 0.574 |
| Amyloidosis | 0 (0.0) | 14 (34.1) | <0.001** |
| Symptoms potentially associated with Fabry disease | |||
| Painful neuropathy | 16 (32) | 11 (26.8) | 0.649 |
| Acroparesthesias | 9 (18) | 4 (9.8) | 0.370 |
| Tinnitus | 6 (12) | 7 (17.1) | 0.556 |
| Hearing loss | 5 (10) | 1 (2.4) | 0.217 |
| Hypohidrosis | 3 (6) | 3 (7.3) | 1.000 |
| Dysmorphic disorder | 0 (0) | 1 (2.4) | 0.451 |
| Treatment | |||
| Anakinra | 0 (0) | 23 (56.1) | <0.001** |
| Canakinumab | 0 (0) | 7 (17.1) | <0.001** |
Continuous data are expressed as medians accompanied by the corresponding minimum and maximum values, while categorical data are presented as counts (n) along with the respective percentages in parentheses.
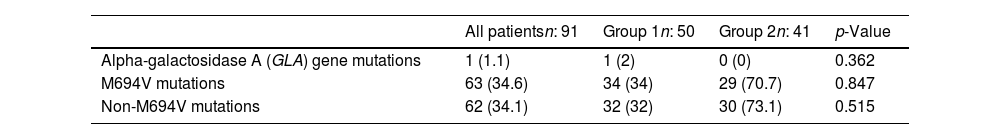
In our exploration of potential genetic factors, a thorough sequence analysis of the entire exons of the GLA gene was conducted for all patients. Notably, a female patient in this group stood out, displaying low GLA enzyme activity (0.1nmol/h/mL) and the presence of the R301Q heterozygous mutation (c.902G>A on exon 6). Intriguingly, the MEFV mutation test yielded negative results for this patient. Her reported symptoms included recurrent abdominal pain, distal extremity pain, and fever during attacks. Additionally, she mentioned experiencing mild acroparesthesia, hypohidrosis, and tinnitus in her detailed medical history. Upon examination of M694V mutations, their occurrence was comparable between the mild and severe groups (34% and 34.6%, respectively), with no statistically significant difference (p=0.847). Similarly, non-M694V mutations demonstrated similarity between the groups, representing a prevalence of 32% in the mild group and 73.1% in the severe group (p=0.515). The specific MEFV mutant alleles and GLA gene mutations identified in FMF patients are summarized in Table 3.
Mutant alleles in MEFV and GLA gene mutations.
| All patientsn: 91 | Group 1n: 50 | Group 2n: 41 | p-Value | |
|---|---|---|---|---|
| Alpha-galactosidase A (GLA) gene mutations | 1 (1.1) | 1 (2) | 0 (0) | 0.362 |
| M694V mutations | 63 (34.6) | 34 (34) | 29 (70.7) | 0.847 |
| Non-M694V mutations | 62 (34.1) | 32 (32) | 30 (73.1) | 0.515 |
Categorical data are presented as counts (n) along with the respective percentages in parentheses.
In this comprehensive study, several key findings emerged, shedding light on the intricate relationship between FMF and FD. Firstly, only one patient (2%) belonging to the mild FMF group tested positive for the FD mutation. Interestingly, none of the individuals in the severe FMF category had abnormal enzyme activity or FD-related mutations. Notable symptoms associated with FD, such as hearing loss, hypohidrosis, acroparesthesia and painful neuropathies, were identified in FMF patients, emphasising the overlapping clinical manifestations of these two conditions. The identification of the R301Q heterozygous mutation related to FD in one FMF patient provided a pivotal validation of our study hypothesis.
Clinically, distinguishing between FD and FMF can be challenging, particularly when patients present with recurrent fever, abdominal pain, and joint pain, without clear indications of a specific systemic disorder.10 This diagnostic challenge is amplified in the presence of a specific ethnic background, often leading clinicians to initially diagnose FMF. However, our study suggests that FD should be suspected, especially in mild FMF cases exhibiting atypical symptoms. The correct diagnosis of FD is important not only for etiologic reasons, but above all to initiate the correct treatment so that patients are not exposed to unnecessary colchicine doses due to FMF diagnosis. Early detection and intervention improve the prognosis of the disease. However, FD is often diagnosed late or incorrectly due to the overlap of the clinical spectrum with other diseases.12 In FD, enzyme replacement therapy has been shown to be the most effective treatment for lowering Gb3 levels prior to long-term deposition.17
FD, a rare X-linked lysosomal storage disease, is characterized by a deficiency of the lysosomal enzyme GLA. The encoding gene, GLA, is situated on the long arm of the X chromosome at position Xq22.18 Over 400 mutations have been identified in the α-Gal A gene, encompassing partial gene deletions, splicing mutations, nonsense mutations, and missense mutations.19 These genetic defects are highly heterogeneous, with most rendering the enzyme nonfunctional. The demonstration of deficient GLA activity in plasma or leukocytes serves as the standard laboratory method for clinically diagnosing FD in males. In contrast, affected females may exhibit normal enzyme activity.20 Consequently, the analysis of the GLA gene to identify mutations responsible for FD becomes pivotal. FD manifests as two major phenotypes. Male FD patients with complete deficiency of α-Gal A activity present severe symptoms, constituting the “severe” FD clinical picture. In contrast, female patients with residual α-Gal A activity display less-evident symptoms, comprising the “mild” FD clinical picture, often emerging at relatively advanced ages.21 The R301Q mutation is reported as one of the mutations associated with the “mild” FD phenotype in various studies. The reported prevalence of FD varies across studies, contingent upon the specific patient cohorts under investigation. In a study by Zizzo et al.,12 exonic mutations in the GLA genes associated with Fabry disease were identified in 9.4% (n=3) of FMF patients with a MEFV gene harboring either a single genetic alteration or no mutation. Lidove et al.22 contributed to this discourse by revealing that among 58 patients diagnosed with Fabry disease, two had prior diagnoses of FMF. Another study by Huzmeli et al.16 unveiled a case where, out of 177 FMF patients with a mutation in the GLA gene, one was diagnosed with FD. In the context of our investigation, approximately 1% of the cases were indicative of FD, underscoring the potential for misdiagnosis as FMF. Notably, our patient, who exhibited the R301Q mutation, belonged to the mild FMF phenotype. These findings underscore the complexity of accurately differentiating between these two conditions and emphasize the necessity for meticulous diagnostic scrutiny, particularly in cases with overlapping clinical features.23–25
The challenge of diagnosing FD persists due to its clinical manifestations, which overlap with those of other pathologies.24 Instances of abdominal pain attacks in FD patients, simulating conditions like appendicitis or renal colic, have been documented. Furthermore, the association of painful attacks with low-grade fever is a recognized feature of FD.10,22 Conversely, clinical manifestations of FMF involve intermittent fever with painful manifestations in the abdomen, chest, and joints.26 The clinical findings of FD, characterized by painful abdominal attacks and fever episodes, can be easily confused with those of FMF, particularly in the absence of other indicative findings of FD.27 Consequently, the analysis of MEFV and GLA genes in suspected FMF cases has proven to be fundamentally important for reducing diagnostic errors.12
The diagnosis of FMF is clinical, based on typical general and site-specific aspects of attacks, as genetic testing has limited sensitivity and specificity.5 In individuals presenting with any of the non-specific symptoms mentioned above, the diagnosis of FD often requires a high degree of suspicion and is based on chemical and genetic testing demonstrating GLA gene mutation and GLA enzyme inactivity. It is crucial to screen patients with FD in populations where there is a high likelihood of detecting occult FD, such as those with undiagnosed renal failure, young patients with cardiovascular disease, and patients with fever of unknown origin, because of the high rate of missed and delayed diagnosis.16,28 Based on knowledge from other high-risk groups, screening patients for FD through exploratory studies is often beneficial. For example, Fancellu et al.29 reported a 41-year-old man identified among 178 young patients with neurovascular disorders. It was discovered that the patient also carried the R227Q GLA mutation, despite initially having only white matter abnormalities. In another study, Paim-Marques et al.30 reported a frequency of FD of 1.12% among 89 patients with juvenile idiopathic arthritis. Other useful investigations of at-risk populations in chronic kidney disease, stroke, cardiac hypertrophy and other populations are frequently reported.
Given the articles highlighting the association between FD and specific clinical scenarios, the emphasis should indeed be on screening FMF patients presenting with the following symptoms; (I) unexplained renal dysfunction or progression despite typical FMF treatment, (II) symptoms that are not entirely consistent with FMF, (III) unexplained left ventricular hypertrophy, arrhythmias or other cardiovascular problems in young FMF patients, (IV) persistent or recurrent fevers that do not fit typical FMF episodes or do not respond to colchicine.12,16,28,31–33 Focusing on these high-risk groups will optimize the use of resources and also increase the likelihood of identifying FMF patients who may have FD. Considering the costs involved and the need for timely diagnosis and treatment, we proposed an algorithm to screen for FD in patients with FMF (Fig. 1).

However, the current study acknowledges a limitation in its relatively small sample size. Despite this constraint, the classification of FMF cases into mild and severe categories and the performance of genetic mutation tests for each patient represent notable strengths of this work.
In conclusion, the striking similarities between the symptoms of FMF and FD pose a diagnostic challenge for physicians, especially in regions where FMF is prevalent. Patients diagnosed with FMF should undergo careful questioning and examination to identify features suggestive of FD, such as acroparesthesia, pain crises, hypohidrosis, tinnitus, and corneal and lenticular opacity. Deficient GLA enzymatic activity and the detection of mutations in the GLA gene can serve as valuable diagnostic indicators in cases where uncertainty exists. Enhancing awareness among clinicians about the intricate interplay of symptoms between FMF and FD is crucial for accurate and timely diagnoses, thereby improving patient outcomes.
Ethical approvalLocal Ethics Committee approval was given by the local medical ethics committee (Decision No: 2020/9) of the Ömer Halisdemir University before starting this study. A signed, informed consent was taken from all participants.
FundingThe author(s) received no financial support for the research, authorship, and/or publication of this article.
Authors’ contributionsSU is the first author of this study. SU is the corresponding author supervising this work. SU, IS drafted the manuscript. IS provided major technical support. TYI, PTK, GK, ZA, NA, SA and FO assisted in the study. All authors contributed to the article and approved the submitted version.
Authors’ statementThe authors hereby confirm that neither the manuscript nor any part of it has been published or is being considered for publication elsewhere. We acknowledge that all authors participated sufficiently in the work and take public responsibility for its content.
Conflicts of interestThe authors have declared no conflicts of interest.


