


Haemorrhagic bullous form of IgA vasculitis (IgAV), or Schönlein-Henoch purpura, is an unusual presentation of the disease in paediatric patients (<2%). Blistering eruptions can sometimes be very striking, leading to hospital admissions and administration of high-dose steroids and even immunosuppressants. Review of the literature, however, does not suggest that this clinical form carries a worse prognosis than the other forms of IgAV. In fact, the prognosis of the disease depends on the organic involvement.
We present the case of a 5-year-old girl that is very representative. She developed palpable purpura and four days later the skin lesions evolved into blistering lesions. She did not receive any anti-inflammatory nor immunosuppressive treatment and the lesions spontaneously subsided within 14 days. She did not develop any extracutaneous nor systemic involvement.
La forma hemorrágico-ampollosa de la vasculitis IgA (VIgA) o púrpura de Schönlein-Henoch, es una de las presentaciones menos frecuentes de la enfermedad en pacientes pediátricos (<2%). Cursa con una afectación cutánea muy llamativa que, con frecuencia, motiva ingresos hospitalarios y tratamiento con corticoides a dosis elevadas, incluso con inmunosupresores. Sin embargo, la revisión de la literatura realizada no sugiere que su pronóstico sea distinto al de otras formas de VIgA, lo que sí parece es que depende de la afectación orgánica existente.
Se presenta el caso de una niña de 5 años que resulta muy representativo. Fue diagnosticada de VIgA hemorrágico-ampollosa, desarrollando lesiones ampollosas 4 días después de la aparición de las lesiones purpúricas. En ningún momento precisó tratamiento antiinflamatorio ni inmunosupresor, resolviéndose las lesiones 14 días después, sin complicaciones significativas.
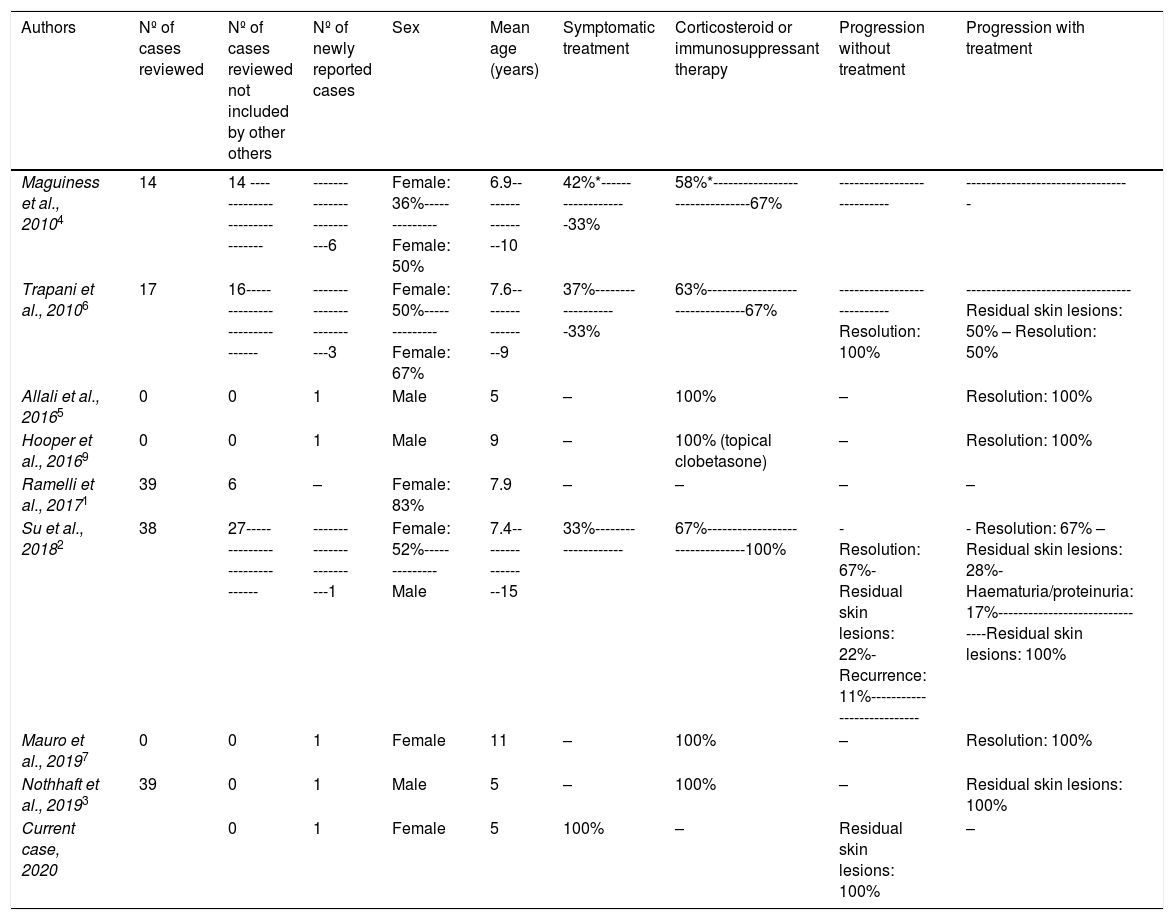
IgA vasculitis (IgAV) or Schönlein-Henoch purpura is the most common vasculitis in infancy.1–7 Palpable purpura predominantly on the lower limbs2–7,9 is the characteristic cutaneous manifestation for diagnosis,8 although it can also present with vesicles and haemorrhagic blisters.4–7 These lesions are much less frequent in children (<2%)1,4,6,9 than in adults, described in fewer than 90 paediatric patients. Therefore, generally no more than one new case is reported per publication (Table 1).
Literature review of paediatric patients diagnosed with haemorrhagic bullous IgA vasculitis.
| Authors | Nº of cases reviewed | Nº of cases reviewed not included by other others | Nº of newly reported cases | Sex | Mean age (years) | Symptomatic treatment | Corticosteroid or immunosuppressant therapy | Progression without treatment | Progression with treatment |
|---|---|---|---|---|---|---|---|---|---|
| Maguiness et al., 20104 | 14 | 14 ----------------------------- | ------------------------6 | Female: 36%--------------Female: 50% | 6.9----------------10 | 42%*-------------------33% | 58%*--------------------------------67% | --------------------------- | --------------------------------- |
| Trapani et al., 20106 | 17 | 16----------------------------- | ------------------------3 | Female: 50%--------------Female: 67% | 7.6----------------9 | 37%-------------------33% | 63%--------------------------------67% | ---------------------------Resolution: 100% | --------------------------------- Residual skin lesions: 50% – Resolution: 50% |
| Allali et al., 20165 | 0 | 0 | 1 | Male | 5 | – | 100% | – | Resolution: 100% |
| Hooper et al., 20169 | 0 | 0 | 1 | Male | 9 | – | 100% (topical clobetasone) | – | Resolution: 100% |
| Ramelli et al., 20171 | 39 | 6 | – | Female: 83% | 7.9 | – | – | – | – |
| Su et al., 20182 | 38 | 27----------------------------- | ------------------------1 | Female: 52%--------------Male | 7.4----------------15 | 33%-------------------- | 67%--------------------------------100% | - Resolution: 67%- Residual skin lesions: 22%- Recurrence: 11%--------------------------- | - Resolution: 67% – Residual skin lesions: 28%- Haematuria/proteinuria: 17%-------------------------------Residual skin lesions: 100% |
| Mauro et al., 20197 | 0 | 0 | 1 | Female | 11 | – | 100% | – | Resolution: 100% |
| Nothhaft et al., 20193 | 39 | 0 | 1 | Male | 5 | – | 100% | – | Residual skin lesions: 100% |
| Current case, 2020 | 0 | 1 | Female | 5 | 100% | – | Residual skin lesions: 100% | – |
It is often ascribed a poor prognosis, and results in high rates of hospitalisation and more aggressive treatment such as IV methylprednisolone and immunosuppressants. Since these lesions can occur in the context of IgAV, without being correlated with poor prognosis,1–4,6 it is important to recognise the condition to differentiate it from other entities with similar presentations but with a worse prognosis.2,6
Clinical observationA healthy 5-year-old girl who, 24 h after onset of fever, cough, and rhinorrhoea, presented with purpuric lesions on the buttocks and lower limbs. She was assessed at her referral hospital where, after taking vital signs and blood (haemogram, coagulation, biochemistry, acute phase reactants) and urine test (urine system), she was diagnosed with IgAV, without complications. On the third day from onset, she began to experience abdominal pain and swelling of the knees and ankles, presenting 24 h later with haemorrhagic blisters. She was assessed at 72 h by paediatric rheumatology, and purpuric lesions and haemorrhagic blisters on the lower limbs were observed (Fig. 1). Her blood pressure and urine system remained unaltered.

She was diagnosed with haemorrhagic bullous IgA vasculitis, and regular blood pressure and urine test strips were taken, which showed no abnormalities during follow-up. She was kept under watchful waiting and did not require corticosteroids or immunosuppressants. The lesions disappeared after 14 days.
The patient was seen for the last time 6 months after the onset of the symptoms and remained asymptomatic, with only a few residual hyperpigmented lesions on the distal area of the lower limbs.
DiscussionHaemorrhagic bullous IgA vasculitis is rare in children, but relatively frequent in adults. It is essential to be aware of its existence to differentiate it from other processes with similar skin lesions that are serious, such as erythema multiforme, toxicoderma (toxic epidermal necrolysis), infections (bullous impetigo, staphylococcal scalded syndrome), autoimmune diseases (pemphigus), and genetic diseases (epidermolysis bullosa).3,9 If there are diagnostic doubts, a skin biopsy can be performed,1 although some authors argue that, with typical clinical symptoms, as in our case, it is not necessary.3,6,9 Characteristically, the blisters appear less than 14 days after the onset of purpura (median 4 days)1 and resolve within 2–4 weeks.1,6 There is no consensus on their treatment,2,5,7 which is not addressed in the European Consensus either,10 unless there is kidney involvement.10 Most authors agree that watchful waiting is a good option,2–4,9 especially in patients in good general condition, as anti-inflammatory treatment helps to control symptoms, but does not seem to reduce the risk of kidney damage, relapse or secuelae.1 In some patients with extensive skin involvement or severe abdominal pain, corticosteroids,2,3 azathiprine,2,3,6 colchicine,3,5 dapsone2,3 and inmunoglobulins7 have been used with variable efficacy.2–4,7 However, these patients appear to have a similar rate of residual skin lesions.2,3 In this regard, up to 25% of patients with blistering forms have been reported to have skin hyperpigmentation, as was the case in our patient, and/or residual scarring,2,3 which is unusual in other forms of IgAV.
ConclusionsThe haemorrhagic bullous form of IgAV is not usually associated with a worse prognosis and does not require different treatment from the usual forms of IgAV.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Alonso de la Hoz J, Martínez Antequera CE, Fernández Manso B, Llorente Otones L, de Inocencio Arocena J. Vasculitis IgA (púrpura de Schönlein-Henoch) hemorrágico-ampollosa, ¿tiene peor pronóstico? Reumatol Clin. 2021;17:549–551.


