




IgG4-related disease is the term used to refer to a condition characterized by a lymphoplasmacytic infiltrate, fibrosis and an increased number of IgG4+ cells present in tissue, in most cases, with an elevated serum IgG4 level. This disease frequently affects the pancreas, salivary glands and lymph nodes, but can involve almost any tissue. Its etiology and the exact role of the different inflammatory cells in the damage to the target organ is still unclear. As yet, there is no international consensus about diagnostic criteria for the disease, but there are important advances in its treatment and in the quest to achieve remission. We include a review of the history, possible pathogenesis, clinical manifestations, diagnostic approach and available therapeutic approaches.
La enfermedad relacionada con IgG4 (ER-IgG4) es una entidad recientemente nominada para definir diversas enfermedades caracterizadas por infiltración linfoplasmocítica, fibrosis, presencia de un número aumentado de células IgG4+ y, en gran parte de los casos, niveles aumentados de IgG4 sérica, afectando frecuentemente el páncreas, las glándulas salivales y los ganglios linfáticos pero pudiendo comprometer casi cualquier estructura de la anatomía humana. Aunque su etiología se desconoce, se han realizado avances en el conocimiento de sus bases fisiopatológicas e inmunológicas, al igual que del rol de las células inflamatorias en el desarrollo de daño del órgano blanco. No existe hasta la fecha un consenso internacional sobre su diagnóstico, lo que no ha impedido avances terapéuticos muy importantes en su control y búsqueda de remisión. Se hace una revisión acerca de la historia, hipótesis sobre la etiología de la enfermedad, sus manifestaciones clínicas, abordaje diagnóstico y terapéutico.
We conducted a nonsystematic review of the literature in English and Spanish in the PubMed and SciELO databases, for the purpose of defining basic aspects of IgG4-related disease (IgG4-RD): the initial historical description of the disease and its course, pathophysiological and immunological bases, clinical manifestations, diagnosis and current therapeutic approach. In both databases, we selected articles published within the last 10 years. In PubMed, we used a search strategy (Immunoglobulin g4 [MeSH] related disease) that yielded a total of 132 results. In SciELO, we used the combination (Igg4 related disease), and found no pertinent results to add to those obtained in PubMed. We did a detailed search for the historical references mentioned in the articles found using the first strategy, as well as the articles of interest mentioned in the respective reference lists. In all, we included 49 references: 42 from the initial search and 7 obtained from the bibliographies.
Introduction and Historical DetailsIgG4-related disease is the name that was given within the last decade to a condition characterized by tumefactive lesions, a dense lymphoplasmacytic infiltrate rich in IgG4-positive cells, storiform fibrosis and, frequently, but not always, elevated serum IgG4 levels.1 The first reports of disease processes compatible with this disorder are from 1892, when Johann von Mikulicz-Radecki described a 42-year-old patient, a farmer, without sicca symptoms, who had “symmetrical edema of the lacrimal, parotid and submandibular glands, with their massive infiltration by mononuclear cells.” He died a year after that description, allowing Dr. Mikulicz to perform the autopsy.2 In it, he confirmed the enlargement of those glands, as well as lymphadenopathy and microscopic evidence of a mononuclear infiltrate. The drawing of what he saw with the microscope is very similar to mucosa-associated lymphoid tissue (MALT) lymphoma but, at the present time, it is impossible to confirm one diagnosis or the other (or the development of the lymphoma in the presence of IgG4-RD), meaning that modern clinicians must always consider malignancy as a differential diagnosis.3
Mikulicz’ syndrome is characterized by swelling of the parotid glands and perhaps the submandibular glands. In 1953, it was included as a possible clinical manifestation of Sjögren's syndrome.4 Years later, in the decade of the 1960s, cases were reported of another condition referred to as “chronic sclerosing pancreatitis”.5 Some of the patients had what was described as a lymphoplasmacytic infiltrate in the affected structure, with a diffuse increase in the size of the organ, but of unknown cause.6 In 1995, the concept of autoimmune pancreatitis was proposed, based on the report of cases of pancreatitis with hypergammaglobulinemia in patients who were autoimmune-positive and responded to corticosteroids.7 However, it was not until 2001, when elevated serum IgG4 levels were recorded in patients with sclerosing pancreatitis, thus separating a new nosological entity.8 In 2004, the documentation of elevated IgG4 concentrations in patients with Mikulicz’ disease established it definitely within the IgG4-RD spectrum.9 The current nomenclature of IgG4-RD was proposed in 2010 and was accepted in 2011 during the first international symposium on this condition, held in Boston. Since then, the annual number of publications on this subject has increased progressively, including, in 2012, the first international consensus on the pathological findings that define it at the present time.10
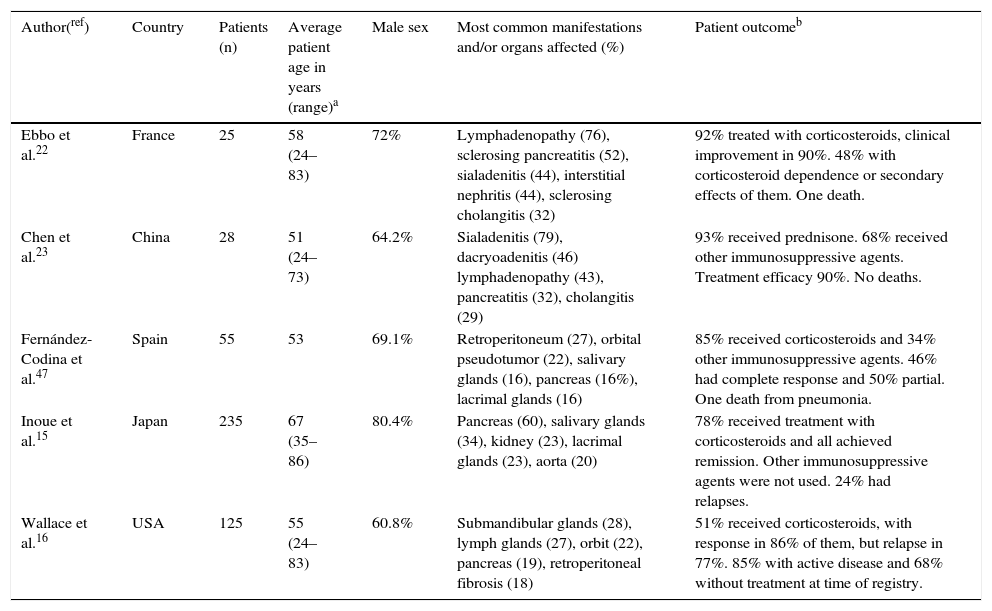
Incidence, Prevalence and Affected OrgansThree histopathological findings characterize the disease in the affected organ: (1) the presence of a storiform pattern of sclerosis; (2) a dense lymphoplasmacytic infiltrate; and (3) an increased proportion of IgG4-positive cells with respect to IgG-positive cells according to immunohistochemical evidence. The average of this proportion is an IgG4+/IgG+ plasma cell ratio >40%, but the criteria varies depending on the organ affected.10 Using these histological findings, it was estimated that the prevalence of IgG4-RD in Japan is 2.63–10.2 cases per million population, with an incidence of 336–1300 new cases each year. To date, the findings in 5 patient cohorts have been published, providing information on the presentation and natural history of the disease (Table 1).
Cohorts Reporting Patients With IgG4-related Disease.
| Author(ref) | Country | Patients (n) | Average patient age in years (range)a | Male sex | Most common manifestations and/or organs affected (%) | Patient outcomeb |
|---|---|---|---|---|---|---|
| Ebbo et al.22 | France | 25 | 58 (24–83) | 72% | Lymphadenopathy (76), sclerosing pancreatitis (52), sialadenitis (44), interstitial nephritis (44), sclerosing cholangitis (32) | 92% treated with corticosteroids, clinical improvement in 90%. 48% with corticosteroid dependence or secondary effects of them. One death. |
| Chen et al.23 | China | 28 | 51 (24–73) | 64.2% | Sialadenitis (79), dacryoadenitis (46) lymphadenopathy (43), pancreatitis (32), cholangitis (29) | 93% received prednisone. 68% received other immunosuppressive agents. Treatment efficacy 90%. No deaths. |
| Fernández-Codina et al.47 | Spain | 55 | 53 | 69.1% | Retroperitoneum (27), orbital pseudotumor (22), salivary glands (16), pancreas (16%), lacrimal glands (16) | 85% received corticosteroids and 34% other immunosuppressive agents. 46% had complete response and 50% partial. One death from pneumonia. |
| Inoue et al.15 | Japan | 235 | 67 (35–86) | 80.4% | Pancreas (60), salivary glands (34), kidney (23), lacrimal glands (23), aorta (20) | 78% received treatment with corticosteroids and all achieved remission. Other immunosuppressive agents were not used. 24% had relapses. |
| Wallace et al.16 | USA | 125 | 55 (24–83) | 60.8% | Submandibular glands (28), lymph glands (27), orbit (22), pancreas (19), retroperitoneal fibrosis (18) | 51% received corticosteroids, with response in 86% of them, but relapse in 77%. 85% with active disease and 68% without treatment at time of registry. |
This condition is generally diagnosed between the sixth and seventh decades of life. It most frequently affects the pancreas, but cases have been reported involving nearly every component of our anatomy, as well as the pediatric population.11–14 Macroscopic and imaging evidence confirm the increase in the size of the affected organ and the presence of fibrosis, a reason why, prior to the era of immunohistochemical diagnosis, different manifestations of the same disease were attributed to different entities and were referred to in terms of the organ involved. Today, Riedel's thyroiditis (fibrous thyroiditis), Küttner tumor (enlargement of the submandibular glands with fibrosis), Ormond's disease (retroperitoneal fibrosis) and Mikulicz’ disease are classified within the IgG4-RD spectrum. Mikulicz’ disease is the prototype of the involvement of organs in this condition, only following the pancreas.
IgG4-related disease occurs predominantly in men. Up to 39% of the patients have a previous or concomitant diagnosis of diabetes mellitus, probably because of the pancreatic involvement.15 In addition to the role of the pancreas, salivary glands and lymph nodes, there are also frequent reports of renal, aortic, retroperitoneal and pulmonary involvement. The variable frequency of these signs in cohorts could be due both to differences in genetic predisposition or risk factors unknown to date, such as differences in the methods employed for diagnosis.15,16 There is also a certain sex-dependent difference in the manifestations (Fig. 1). One of the main conclusions of the cohort analysis is that the involvement of a single organ is the exception, rather than the rule in IgG4-RD, which is a multisystem disease in most cases.16

Percentage of patients with certain organs affected by confirmed IgG4-related disease.
Source: Adapted from Hasosah et al.14
In contrast to other types of pancreatitis, that provoked by IgG4-RD is not related to the presence of circulating autoantibodies. It is generally accompanied by abdominal pain, jaundice secondary to edema and pancreatic and bile duct infiltration, weight loss and exocrine or endocrine pancreatic insufficiency.17 Renal involvement is more varied, and there are reports of tubulointerstitial nephritis, pain due to kidney enlargement, varying degrees of hematuria and proteinuria or obstructive nephropathy secondary to retroperitoneal fibrosis.18 In pulmonary involvement, almost any radiological pattern can be observed, including: (1) localized parenchymal involvement; (2) diffuse parenchymal involvement; (3) presence of lymphadenopathy; (4) diffuse interstitial pattern; (5) bronchial wall thickening; and (6) pulmonary nodules. Given the heterogeneity of lung involvement, it sometimes can only be confirmed by biopsy.19 Fibrotic processes in can also be viewed in imaging studies in neck, mediastinum and abdomen.20,21 Neoplasms have been reported to occur simultaneously or develop later on in patients with a diagnosis of IgG4-RD. Of those diagnosed at the same time, gastric, colorectal and prostate neoplasms are those most commonly encountered and, of those discovered subsequently, lung neoplasm is the disorder most frequently found, although, to date, a cause/effect relationship between IgG4-RD and malignant disease has not been identified.15
Diagnosis of the DiseaseThe presentation of IgG4-related disease may have limited specificity or suggest other much more common conditions. In fact, on occasion, it may be accompanied by other inflammatory processes, such as autoimmune diseases, systemic vasculitides or neoplastic disorders, but we still cannot point to the pathophysiological bases for this association.
In a French cohort, in a third of the cases, the patients were mostly concerned about constitutional symptoms, weight loss and sicca symptoms. Physical examination revealed lymphadenopathy in nearly half of them, but only one fifth had jaundice or enlarged salivary glands.22 In a Chinese cohort, salivary gland swelling was the predominant symptom or finding, followed by a history of allergic disease in half of the patients, abdominal pain and lymphadenopathy.23
Imaging studies revealed an enlargement of the affected organ or regional fibrosis. Positron emission tomography (PET), which is useful in hypermetabolic lesions (of inflammatory nature, not infectious or neoplastic), enables the detection of increased metabolic activity in the affected organs, but does not differentiate between the distinct etiologies with this pattern. Its utility would be in documenting multiorgan involvement.24,25 Among the laboratory findings, it is not rare to observe elevated serum levels of total IgG (61% >1800mg/dL), IgG4 (84% >135mg/dL) and IgE (58% >360mg/dL). The incidence of autoantibodies is similar to that expected for the age group in which it develops.26
Although IgG4 has lent its name to the disease, the serum levels of this antibody are not elevated in all of the patients in whom the condition has been diagnosed by biopsy. According to the inclusion criteria employed in the study, the sensitivity of serum IgG4 ranges between 50% and 90%, with a highly variable specificity that can be as low as 60%.27,28 An elevated IgG4 level can be found in a number of respiratory disorders (bronchiectasis, asthma, chronic sinusitis, sarcoidosis), biliary diseases (primary sclerosing cholangitis, cholangiocarcinoma, lithiasis), chronic pancreatitis of other etiologies and cirrhosis. Among the autoimmune diseases, Sjögren's syndrome, systemic lupus erythematosus, rheumatoid arthritis, inflammatory myopathies and vasculitides may also be associated with a high serum concentration of this immunoglobulin.26,29
There is no internationally accepted criteria for the diagnosis of IgG4-RD. Japan has taken the lead in research and information about the disease, generating at least 2 groups of diagnostic criteria (Table 2). On the other hand, it may be the sum of the clinical criteria, as well as the histopathology and serology, that defines the probability that a given patient has the disease, which can be confirmed when all 3 are met and ruled out in their absence30 (Fig. 2). Clinicians should let themselves be guided by the reported clinical signs, radiological evidence of fibrosis, enlargement of organs or lymphadenopathy, elevated serum IgG4 levels in the absence of some other explanation and the characteristic pathological findings in conventional and immunohistochemical staining. The means should always be sought to perform the biopsy in such a way as to rule out differential diagnoses.31 As was mentioned above, it is much easier to encounter a multiorgan disease than conditions involving a single organ. Therefore, the study should be systematic and guided by the clinical signs and laboratory tests.
Two Criterion Groups for Diagnosis of IgG4-related Disease.
| Criteria of Umehara et al.12 | Criteria of Okazaki et al.48,49 |
|---|---|
| Highly suggestive clinical findings Symmetrical edema of lacrimal, parotid or submandibular glands, autoimmune pancreatitis, inflammatory pseudotumor, retroperitoneal fibrosis, suspected Castleman's disease Highly suggestive laboratory findings IgG4>135mg/dL IgG4+ cells>40% of total IgG+ cells Suggestive clinical findings Unilateral lacrimal, parotid or submandibular gland edema, orbital pseudotumor, sclerosing cholangitis, prostatitis, hypertrophic pachymeningitis, interstitial pneumonitis, interstitial nephritis, thyroid changes, hypophysitis, inflammatory aneurysm Suggestive laboratory findings Unexplained hypergammaglobulinemia, hypocomplementemia, hyper IgE syndrome or eosinophilia, lymphadenopathy according to nuclear medicine | 1. Enlargement or focal or diffuse lesions in one or more organs 2. Serum IgG4 concentrations>135mg/dL 3. Histopathology (a) Lymphocytic and plasma cell infiltrate with fibrosis, without neutrophilic infiltrate (b) IgG4+ plasma cell infiltrate greater than 10 per high-power field or proportion of IgG4/IgG>40% (c) Storiform whorled fibrosis (d) Obliterative phlebitis The diagnosis is based on meeting one or more of the following combinations of criteria: • 1+2 • 1+3 (a+b) • 2+3 (a+b) • 3 (a+b+c+d) |

Evidence of the diagnosis of IgG4-related disease according to diagnostic criteria.
Source: Adapted from Ryu et al.29
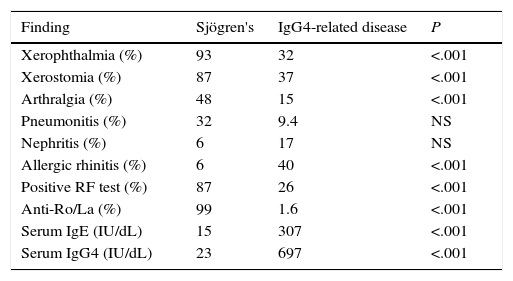
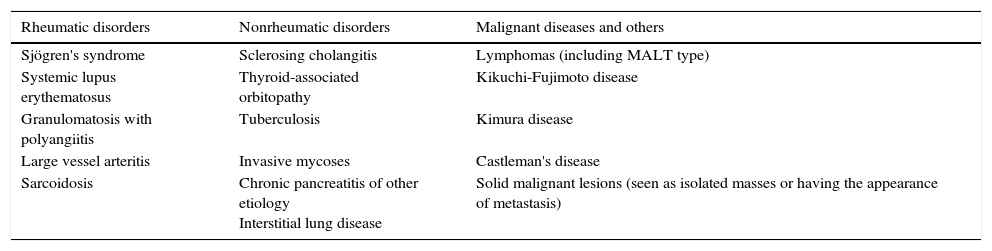
Given that one of the main differential diagnoses of this disease is Sjögren's syndrome, the clinician should keep in mind certain aspects that make it possible to distinguish the two diseases. In contrast to IgG4-RD, Sjögren's syndrome is more common in women, it is not usually associated with swelling of the lacrimal glands, it begins with sicca symptoms in a great many cases, it usually generates gland destruction by means of inflammation and it has a clearly defined serological profile of autoimmunity. In histopathology, the criteria for the classification of Sjögren's syndrome requires a focus score of at least 1, defined as the presence of at least 1 minimal focus of 50 lymphocytes adjacent to gland acini of normal aspect for every 4mm2 of tissue, a finding that is very different from what has been mentioned for IgG4-RD.32 Other useful clinical and laboratory data are shown in Table 3. The complete list of differential diagnoses is extensive and includes a number of diseases with multisystem involvement, generalized lymphadenopathy and nonspecific findings in terms of the diagnosis. The most common ones can be seen in Table 4.
Clinical and Laboratory Differences Between Sjögren's Syndrome and IgG4-related Disease.
| Finding | Sjögren's | IgG4-related disease | P |
|---|---|---|---|
| Xerophthalmia (%) | 93 | 32 | <.001 |
| Xerostomia (%) | 87 | 37 | <.001 |
| Arthralgia (%) | 48 | 15 | <.001 |
| Pneumonitis (%) | 32 | 9.4 | NS |
| Nephritis (%) | 6 | 17 | NS |
| Allergic rhinitis (%) | 6 | 40 | <.001 |
| Positive RF test (%) | 87 | 26 | <.001 |
| Anti-Ro/La (%) | 99 | 1.6 | <.001 |
| Serum IgE (IU/dL) | 15 | 307 | <.001 |
| Serum IgG4 (IU/dL) | 23 | 697 | <.001 |
NS: no significant; RF, rheumatoid factor.
Source: adapted from Masaki et al.50
Disorders Frequently Included in the Differential Diagnosis of IgG4-related Disease.
| Rheumatic disorders | Nonrheumatic disorders | Malignant diseases and others |
|---|---|---|
| Sjögren's syndrome | Sclerosing cholangitis | Lymphomas (including MALT type) |
| Systemic lupus erythematosus | Thyroid-associated orbitopathy | Kikuchi-Fujimoto disease |
| Granulomatosis with polyangiitis | Tuberculosis | Kimura disease |
| Large vessel arteritis | Invasive mycoses | Castleman's disease |
| Sarcoidosis | Chronic pancreatitis of other etiology Interstitial lung disease | Solid malignant lesions (seen as isolated masses or having the appearance of metastasis) |
MALT, mucosa-associated lymphoid tissue.
Under normal conditions, IgG4 constitutes less than 5% of total IgG, and is the least frequent subtype; however, its relative count is elevated by up to 80% in chronic allergies.18 Its structure, like that of the other subtypes, consists in 2 heavy and 2 light chains, but the disulfide bridges that join the 2 heavy chains are unstable, and allows them to separate and mix with other IgG4 fragments, thus generating divalent molecules.33 This immunoglobulin has a poor capacity for activating complement and forming immune complexes. Therefore, it has been postulated that it has an anti-inflammatory role as it competitively blocks other much more active subtypes.34,35 Nevertheless, it has been implicated in another 3 diseases: pemphigus foliaceus (in which the subtype predominates in subepidermal deposits), thrombotic thrombocytopenia purpura (in which there are increased levels of circulating IgG4) and certain cases of membranous glomerulonephritis.1 Its role as a cause of fibrosis in IgG4-RD is not that clear. A model of the pathogenesis of the disease shows that some unknown factor (a viral infection or unidentified antigen) triggers an immune response, provoking an infiltration of the organ by B cells that differentiate into plasma cells. In the specific case of the pancreas, the choice of IgG4+ cells would be made by the myofibroblasts (stellate cells). This model explains infiltration, but not tissue fibrosis.36 Tissue infiltration is attributed not only to plasma and B cells, but there is a large number of CD4 cells. These cells and their mediators (interleukins, tumor necrosis factor) could stimulate the proliferation of fibrous tissue, but the cause of the stimulus to CD4 cells is not clarified either. Carbonic anhydrase II and IV or certain pancreatic enzymes could serve as antigens for organ infiltration by inflammatory cells, but these are not found in all the tissues that can be affected by the disease.37 The profile of circulating cytokines is usually T helper 2 (Th2) cells, thus explaining the increase in serum and bound IgG4, but the amount of Th2 cells is not affected by treatment with corticosteroids, regardless of the clinical response.38 Despite the knowledge acquired, there are still questions to be solved: What autoantigen, if there is one, triggers the disease? What is the exact mechanism of tissue fibrosis? Does bound IgG4 play any role?
Treatment of IgG4-related Disease and Patient Follow-upInternational consensus has recently been achieved concerning the treatment of the disease (although this does not exist with regard to the diagnosis). A clinician should first ask whether a patient needs treatment, and the answer will be affirmative in a large majority of cases. Arguments in favor include a better response and prognosis in terms of the affected organ with early initiation of therapy, plus the possibility of a case of multiorgan involvement also benefiting from the treatment and the generally favorable response to it. Exceptions would include patients with disease having minimal symptoms or with no prognostic implications (enlargement of salivary or lacrimal glands or lymphadenopathy that is isolated and not restrictive), in which the involvement of other organs has been ruled out. Others might be patients with a predominantly fibrous reaction (orbital pseudotumors, sclerosing mesenteritis), who would benefit most from surgical intervention than from medical therapy.31 It should be taken into account that the disease is unpredictable in terms of the organs it affects and that patients with induced or spontaneous remission may have relapses in other systems.39 Moreover, a delay in treatment for any reason can result in serious and irreversible sequelae.40
IgG4-related disease usually responds to high-dose glucocorticoids, with an average dose of 40mg/day, which is subsequently tapered.31 The definition of “response” to treatment differs depending on the bibliography, and there is even a scale designed to evaluate it. However, in practice, it is contingent upon the criteria of the attending physician.41 In this disease, relapsing is quite frequent. In a cohort of 116 patients with type 1 autoimmune pancreatitis (a manifestation of IgG4-RD), the initial dose of prednisolone was 40mg, with a gradual decrease of 5mg each week until it was discontinued. Half of the patients had a relapse of the disease.42 International consensus recommends a much more gradual tapering of the dose, over a period of 3–6 months. With this dosing scheme, the rate of relapses was 23% in this cohort from the United States.16 In their cohort, the Japanese maintained corticosteroids at low doses (2.5–5mg/day) without using steroid-sparing agents for 3 years, with a reported relapse rate of 24%.15 The treatment proposed for initial relapses was a new course of corticosteroids similar to the first.
With respect to the use of steroid sparing agents, there are 3 alternatives: to use them from the start together with the corticosteroids, begin them at the time of the first relapse or not employ them at all. In the cohort of patients with autoimmune pancreatitis mentioned above, treatment after the first relapse was azathioprine, 6-mercaptopurine or mycophenolate mofetil, and there were no differences in the time until a second relapse with respect to patients who received another course of corticosteroids (70% vs 60% at 48 months).42 Treatment with the 3 medications was discontinued due to adverse effects in 17%, 33% and 0% of the patients, respectively. In the cohort from the United States, only 1 patient received a steroid-sparing agent,16 whereas in the Japanese cohort, none of them did, with the results mentioned above. In the international consensus statement, only 46% of the experts consulted agreed with the use of one of these 3 agents at some time of treatment, with no specific recommendations with respect to their indication.
Very favorable results have been published concerning the use of rituximab (RTX) as induction and/or maintenance therapy. A recent study evaluated the efficacy of RTX in 12 patients at a dose of 375mg/m2 weekly for 4 weeks, followed by new doses in accordance with the criteria of the attending physician. The indication for its use was the discontinuation due to therapeutic failure of steroid sparing agents or for corticosteroid intolerance. Complete remission was defined as clinical, radiographic and biochemical resolution in terms of the pancreas or affected organ, absence of new inflammatory lesions during follow-up and discontinuation of maintenance therapy to control the disease. Partial remission was the presence of an improvement in the patient, although resolution of the inflammation (clinical, radiographic and biochemical) without the need for corticosteroids had not been achieved, and incomplete remission was the attainment of improvement in the inflammatory changes, but requiring concomitant therapy with RTX and corticosteroids. Ten (83%) of the 12 patients achieved complete remission, one had partial remission and another, incomplete remission.42
In another study, RTX was used as induction therapy at a dose of 1000mg on days 1 and 15, without simultaneous corticosteroids (26 patients) or with a gradual reduction of the dose of the latter during the first 2 months (4 patients). Using a responder index to measure IgG4-RD activity, 97% of the individuals had an activity response at 6 months, with 47% in complete remission, and 40% maintaining that response at 12 months.43 The initial dose of RTX seems to be of great importance, given that, in another study, a single dose of 500mg of RTX with a scheme requiring the tapering of the corticosteroid dose, was associated with a high number of relapses and absence of a response.44 Methotrexate has also been employed as maintenance therapy together with corticosteroids, with acceptable results (50% of disease remission at 24 months), but with no comparison group or other study to establish conclusions on the use of this agent in IgG4-RD.45
Follow-up of the patients is essentially clinical, with the aid of the conventional laboratory tests, depending on the organ affected, aside from imaging techniques.31 It has also been proposed that the serum IgG4 levels be monitored, as they decrease once treatment has begun, and any subsequent increase seems to be correlated with relapses.44,46 The number of circulating plasmablasts and serum CD19 levels were employed on an experimental basis, but their use and utility in clinical practice will need to be standardized.38
ConclusionsIgG4-RD is a recently described condition whose etiology and pathophysiology have yet to be clarified. It is characterized by a fibrotic pathology and lymphoplasmacytic infiltrate with a predominance of IgG4+ cells in one or several target organs. Those most frequently involved are the pancreas, lymph nodes and salivary glands, but it can affect most of the structures of our anatomy, and the majority of the patients have elevated serum IgG4 levels. Despite the name, there has been no clear definition of the role of IgG4 in this disorder, of the stimulus that provokes infiltration of an organ by inflammatory cells, or of factors that determine which organ or organs will become involved in the disease. As there is no reliable diagnostic criteria, it is the sum of arguments (clinical presentation, imaging studies, elevated serum IgG4, histopathology, response to corticosteroids) that leads to the diagnosis. At the present time, its treatment consists of systemic corticosteroids at high doses, that are subsequently tapered. There is currently no evidence supporting the use of steroid sparing agents, but biological therapy with RTX has been employed successfully in the induction and maintenance of the clinical response, and has been proposed as second-line therapy.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
FundingJose A. Gómez-Puerta has a grant from Colciencias (656/2014).
AuthorshipOAS and JAGP participated in the conception, design, analysis and interpretation of the data, and in writing, reviewing and approving the submitted manuscript. AA participated in the analysis and interpretation of the data, and in writing, reviewing and approving the submitted manuscript.
Conflicts of InterestThe authors declare they have no conflicts of interest.
Please cite this article as: Ardila-Suarez O, Abril A, Gómez-Puerta JA. Enfermedad relacionada con IgG4: revisión concisa de la literatura. Reumatol Clin. 2017;13:160–166.


