





We report the case of a 50-year-old female smoker with an 11-year history of seropositive rheumatoid arthritis (rheumatoid factor and anti-cyclic citrullinated peptide antibodies) receiving triple therapy. She developed pulmonary nodules diagnosed as Langerhans cell histiocytosis by lung biopsy. We found no reported cases of the coexistence of these two diseases. Smoking abstinence led to radiologic resolution without modifying the immunosuppressive therapy.
Se presenta el caso de una mujer de 50 años, fumadora, con artritis reumatoide seropositiva (FR y CCP) de 11 años de evolución en tratamiento con triple terapia, y aparición de nódulos pulmonares con diagnóstico final de histiocitosis de células de Langerhans por biopsia pulmonar. No hemos encontrado casos descritos de la coexistencia de ambas enfermedades. La abstinencia tabáquica llevó a la resolución radiológica sin necesidad de modificar la terapia inmunosupresora.
The patient was a 50-year-old woman who smoked 20 cigarettes/day. She had been diagnosed with seropositive (rheumatoid factor [RF] and anti-cyclic citrullinated peptide antibodies [anti-CCP]), erosive rheumatoid arthritis (RA) in 1999. She was being treated with methotrexate (MTX) since 2000, in combination with salazopyrin and hydroxychloroquine since February 2009, at which time, she had achieved complete remission. In March 2010, in relation to a self-limiting case of a cold, her primary care physician had asked for a chest radiograph, which showed possible images of nodules, predominantly in the upper lobes. The patient was asymptomatic and the physical examination was normal. She had an immunological study in which she tested positive for RF and anti-CCP and negative for antinuclear antibodies and antineutrophil cytoplasmic antibodies; the Mantoux text was positive, and chest computed tomography revealed multiple bilateral pulmonary nodules measuring about 0.5cm, some showing cavitation, especially in images from the upper and middle lobes, but the lower lobes were also involved (Fig. 1A). The bronchoscopy was normal, with negative results in the smear, bronchoalveolar lavage fluid culture and cytology of the bronchial aspirate for malignant cells. The patient was referred to undergo lung biopsy. Videothoracoscopy revealed that the lung parenchyma had small scattered subpleural lesions, and the pathological study showed the presence of foamy histiocytes with grooved nuclei, with other multinucleated and somewhat elongated histiocytes occupying the alveolar spaces (Fig. 1C). Immunohistochemical techniques identified cells that were positive for CD1a (Fig. 1D) and for S100, as well as langerin in the histiocytes described, all of which was consistent with Langerhans cell (LC) histiocytosis (LCH). At that time, it was recommended that she quit smoking and the same immunosuppressive therapy was maintained. During that entire period, she had no joint or respiratory symptoms and underwent radiographic follow-up 6 months later. The images showed residual pulmonary cysts and the nodules had disappeared (Fig. 1B). After 5 years of follow-up with no new incidences, the patient continues to receive triple immunosuppressive therapy.
Discussion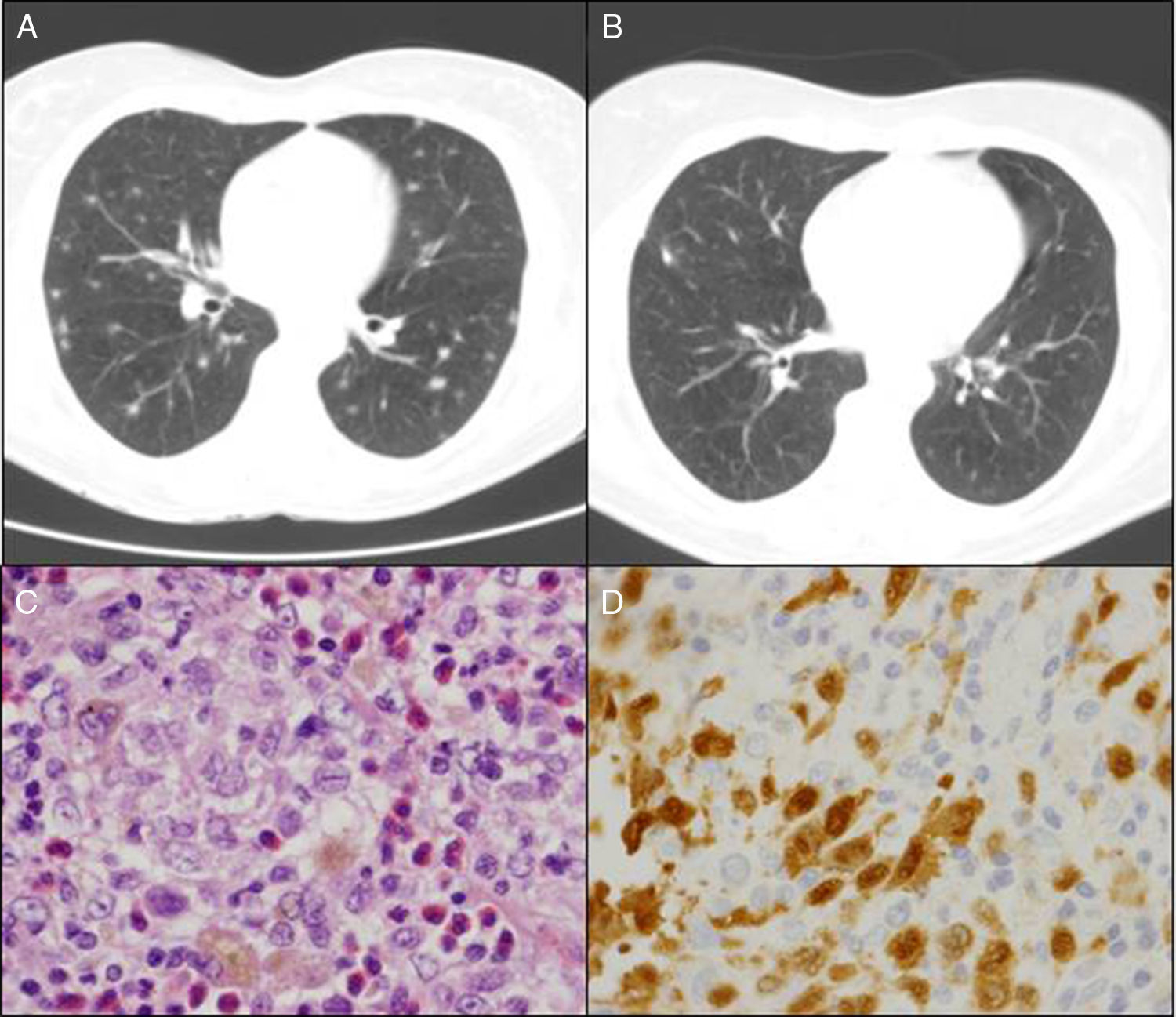
Histiocytes are cells of the immune system that include both macrophages and dendritic cells (non-macrophage antigen-presenting cells). Histiocytoses are rare diseases, LCH being the most representative, characterized by the infiltration of LC, a type of dendritic cell found predominantly in the pulmonary alveoli and in the skin, with its distinctive “racket”-shaped cytoplasmic Birbeck granules. The term LCH was coined in the attempt to confer a better classification and identification of the patients, as it combines previous entities (eosinophilic granuloma, histiocytosis X, etc.) in which the lesions were due to a proliferation and infiltration of the same cell type, the identification of LC being the diagnostic criteria called for since then.1 The pathogenesis is unknown, there being either a reactive or clonal proliferative response, with different degrees of phenotypic aggressiveness in the infiltrated organs or systems (Table 1). Pulmonary Langerhans cell histiocytosis (PLCH) is the most representative form in adults and is usually recognized as a separate entity.
Classification of Histiocytosis.
| Simplified classification of histiocytosisa |
|---|
| 1. Langerhans cell histiocytosis (LCH) |
| 2. Hemophagocytic lymphohistiocytosis (HLH) |
| 3. The rare histiocytic disorders (RHD) |
| Juvenile xanthogranuloma |
| Erdheim-Chester disease |
| Multicentric reticulohistiocytosis |
| Rosai-Dorfman disease |
| The malignant histiocytosis |
| Classification of Langerhans cell histiocytosis |
|---|
| 1. Isolated disease in a single organ or systemb |
| Pulmonary LCH (85% of the cases of pulmonary LCH in adults) |
| Bone (single or multiple) |
| Skin/hypothalamus/hypophysis/lymph nodes/liver, spleen, thyroid glands |
| 2. Multisystem diseasec: affecting two or more organs |
This disorder occurs in young adult smokers, and smoking cessation can lead to partial or total remission of the pulmonary lesions. In the early stages, it is characterized by bronchoalveolar inflammatory changes and, in the most advanced phases, by cystic lung destruction. The clinical manifestations vary widely: dyspnea, cough, weakness, fever, weight loss, pleuritic chest pain and, occasionally, spontaneous pneumothorax. The incidence of hemoptysis is minor; thus, should it occur, the cause should be looked for elsewhere (mainly tumor-related). The disease can be detected incidentally in radiological studies performed in asymptomatic patients, occurring in around 15% of the cases, and the same proportion can be applied to those with extrapulmonary manifestations, such as bone cysts, diabetes insipidus or exanthema.2,3 Computed tomography is the technique of choice to characterize pulmonary involvement. The combination of multiple cysts and bilateral nodules distributed throughout the medium and upper lobes, with or without interstitial thickening, in a young smoker, is considered to be a characteristic reason to strongly suspect PLCH.4 This was not the case in our patient, whose disorder had to do mainly with the nodules, there being few cystic lesions and involvement of the lower lobes. Bronchoalveolar lavage is useful for the diagnosis when more than 5% of the LC are detected by electron microscopy or immunostaining, but this technique is not available at every medical center.5 The combination of the typical radiological findings, together with the positive results of bronchoalveolar lavage, can be accepted as a sufficient diagnostic criteria, without pathological confirmation.2 However, should the latter be necessary, the approach should be pulmonary biopsy, since transbronchial biopsy has been found to have a very limited diagnostic yield. The presence of LC is confirmed by immunohistochemical techniques with monoclonal antibodies against the CD1a membrane antigen, the intracellular S100 protein and langerin.6
The differential diagnosis should mainly include the formal screening for mycobacteria, hematogenous infections, other types of nodulosis such as sarcoidosis, silicosis, vasculitis, metastases or primary pulmonary tumors, and in our case, obviously, with pulmonary rheumatoid nodules (RN). With respect to the latter, we must recall that pulmonary RN are classically related to tobacco use, RF positivity, coexistence of cutaneous RN and human leukocyte antigen (HLA) DRB1, although there are also cases induced by treatment with synthetic disease-modifying antirheumatic drugs or biological therapy with anti-tumor necrosis factor (TNF) agents. Rheumatoid nodulosis has long been known to be induced by MTX. It is characterized by the rapid development of RN that are histopathologically similar to classic RN, that stops once the drug is no longer being administered and reappears if the attempt is made to reintroduce it.7 It has been associated with the HLA DRB1*0401 allele. Pulmonary rheumatoid nodulosis of similar temporal characteristics as that induced by MTX is much less common. In contrast, in other situations in which the development of cutaneous or pulmonary RN during prolonged chronic treatment with MTX, the causal association would be much more questionable. Moreover, on most occasions, there is a characteristic disparity between the development of nodulosis and a good control of the joint disease. Thus, the strategy to be followed with respect to discontinuing treatment can also be debated, and this all points to differences between the pathophysiological mechanisms that result in the formation of rheumatoid granulomas and those that lead to synovitis and synovial hypertrophy. Similar problems with interpretation in the literature can be found with leflunomide and anti-TNF therapy as inducers of pulmonary rheumatoid nodulosis.8 Finally, the differential diagnosis of purely cystic PLCH would include centrilobular emphysema, cystic fibrosis, tuberous sclerosis and, specifically in women, lymphangioleiomyomatosis (Table 2).
Differential Diagnosis of Pulmonary Nodules in a Patient With Pulmonary Langerhans Cell Histiocytosis and Rheumatoid Arthritis.
| Tumor-related | |
| Lung tumor | |
| Metastasis | |
| Lymphangitic carcinomatosis | |
| Infectious | |
| Hematogenous infection: Staphylococcus aureus, Klebsiella pneumoniae, Pseudomonas | |
| Granulomatosis: mycobacterial, Nocardia, fungi | |
| Parasites: hydatid disease, paragonimiasis, Echinococcus | |
| Inflammatory | |
| Sarcoidosis | |
| Pulmonary Langerhans cell histiocytosis | |
| Pneumoconiosis: asbestosis, silicosis, Caplan syndrome | |
| Rheumatoid nodules | |
| Vasculitis: granulomatosis with polyangiitis, eosinophilic granulomatosis | |
| Cryptogenic organizing pneumonia | |
| Drug-related | |
| Rheumatoid nodules induced by disease-modifying antirheumatic drugs (methotrexate and leflunomide) and anti-tumor necrosis factor agents | |
| Amiodarone, bleomycin, carbamazepine, others | |
| Miscellaneous | Differential diagnosis of the cystic pattern on radiology |
| Respiratory bronchiolitis | Pulmonary Langerhans cell histiocytosis |
| Amyloidosis | Lymphangioleiomyomatosis |
| Pulmonary embolism | Cystic fibrosis |
| Congenital pulmonary airway malformation | Tuberous sclerosis |
The natural course of PLCH is unpredictable; spontaneous remission can be estimated to occur in up to 50% cases, and severe progressive respiratory failure can develop in 10%–20% of the patients. The major consideration for the treatment of PLCH is smoking cessation, which may be achieved in up to one third of the cases. This simultaneously eliminates an important risk factor for lung cancer, chronic obstructive pulmonary disease and cardiovascular disease. However, it is estimated that the disease continues to progress in as many as one third of the patients who stop smoking. Cases of recurrence have been reported in those who quit. The use of corticosteroids as monotherapy or in combination with immunosuppressive agents (methotrexate, cyclophosphamide, vinblastine and, more recently, cladribine) is questioned because of the absence of controlled clinical trials and due to the uncertainty regarding the natural behavior of the disease. It is usually reserved for cases of functional deterioration revealed by spirometry, progressive interstitial disease or extrapulmonary multisystemic involvement. Overall 5-year survival is estimated to be 75%, and the factors most widely associated with poor prognosis are advanced age and deterioration in respiratory function tests and its extension at the time of the diagnosis.3 Lung transplantation is an option for long-standing severe disease, although recurrences have been reported in the transplanted organ.9
Our patient was in clinical remission for her joint disorder, and was receiving triple therapy when the pulmonary manifestations were detected. We maintained the same strategy, especially after seeing the radiological improvement once she had achieved smoking cessation. We found no cases in the literature on the previous coincidence of PLCH and RA. There is a recently described case of PLCH in a patient with Sjögren's syndrome treated with oral steroids and azathioprine, with a favorable radiological and functional response.10 Multicentric reticulohistiocytosis may be a cause of erosive arthritis and has been associated in patients with RA and Sjögren's syndrome,11 although clinically, it is a differential diagnosis that was not considered in our case. With respect to the attitude to take if at some time, she were to need biological therapy, we have only had access to the favorable communication of 2001 on the part of Henter et al., in reference to a patient with very aggressive multifocal infantile LCH with a good response to etanercept.12 Since then, communications along those lines have not been repeated.
In short, PLCH is the most representative type of LCH in adulthood. It is directly related to tobacco use, and smoking cessation leads to a high percentage of cases in which the outcome proved to be the solution to the disease. We have not found previous references in the literature to the simultaneous coexistence of RA and PLCH. Our patient did not have a characteristic radiological pattern, and triple immunosuppressive therapy does not appear to have influenced the favorable change in the symptoms after more than 5 years of follow-up after smoking cessation.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of InterestThe authors declare they have no conflicts of interest.
Please cite this article as: Zurita Prada PA, Urrego Laurín CL, Assyaaton Bobo S, Faré García R, Estrada Trigueros G, Gallardo Romero JM, et al. Artritis reumatoide y nódulos pulmonares: un diagnóstico final inesperado. 2017;13:167–170.


