




La asociación entre lupus eritematoso sistémico (LES) y púrpura trombótica trombocitopénica (PTT) es una situación descrita pero poco frecuente y suele ocurrir en casos con actividad lúpica y deterioro renal. Presentamos un caso de PTT con disminución de los niveles de ADAMST13 que ocurrió en una paciente afectada de LES pero sin actividad y sin afectación renal. Fue un caso que presentó dificultades diagnósticas iniciales y que, debido a su refractariedad, precisó recambios plasmáticos y tratamientos inmunosupresores, incluyendo rituximab.
The association between systemic lupus erythematosus (SLE) and thrombotic thrombocytopenic purpura (TTP) has been infrequently reported. Usually, patients with TTP have more SLE activity and frequent renal involvement. Here we present a case of TTP associated to low-activity SLE. The absence of renal and major organ involvement increased the difficulty in making the initial diagnosis. ADAMTS13 activity in plasma in this patient was very low, as seen in other similar cases. The evolution of the patient was poor, needing plasma exchanges and immunosuppressive therapy, including the use of rituximab.
La púrpura trombótica trombocitopénica (PTT) se caracteriza por la presencia de fiebre, anemia hemolítica microangiopática, trombocitopenia y alteraciones neurológicas y renales1,2. Aunque la mayoría de los casos son idiopáticos, existen casos familiares y casos que se asocian a neoplasias, infecciones, fármacos, embarazo y colagenosis3. La PTT idiopática se debe a la presencia de anticuerpos inhibidores frente a la metaloproteasa ADAMTS132,4,5.
La PTT es una entidad grave con elevado índice de mortalidad si no se trata. El tratamiento con recambios plasmáticos ha mejorado mucho el pronóstico y la mortalidad ha disminuido desde el 85-100% hasta el 10-30% actual2,5,6. Sin embargo, existen casos refractarios y recaídas en 40% de los pacientes. En estas situaciones se han empleado varios agentes inmunosupresores, entre ellos rituximab (RTX)7,8.
La PTT es una complicación infrecuente en el contexto del lupus eritematoso sistémico (LES). Los casos descritos en la literatura médica se producen habitualmente en pacientes con actividad lúpica intensa y con afectación renal9. Presentamos a continuación un caso de LES que, estando con buen control de la actividad lúpica, desarrolló de forma brusca un episodio de PTT de curso agresivo y refractario a recambios plasmáticos, requiriendo tratamiento inmunosupresor adicional, incluyendo RTX.
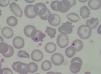
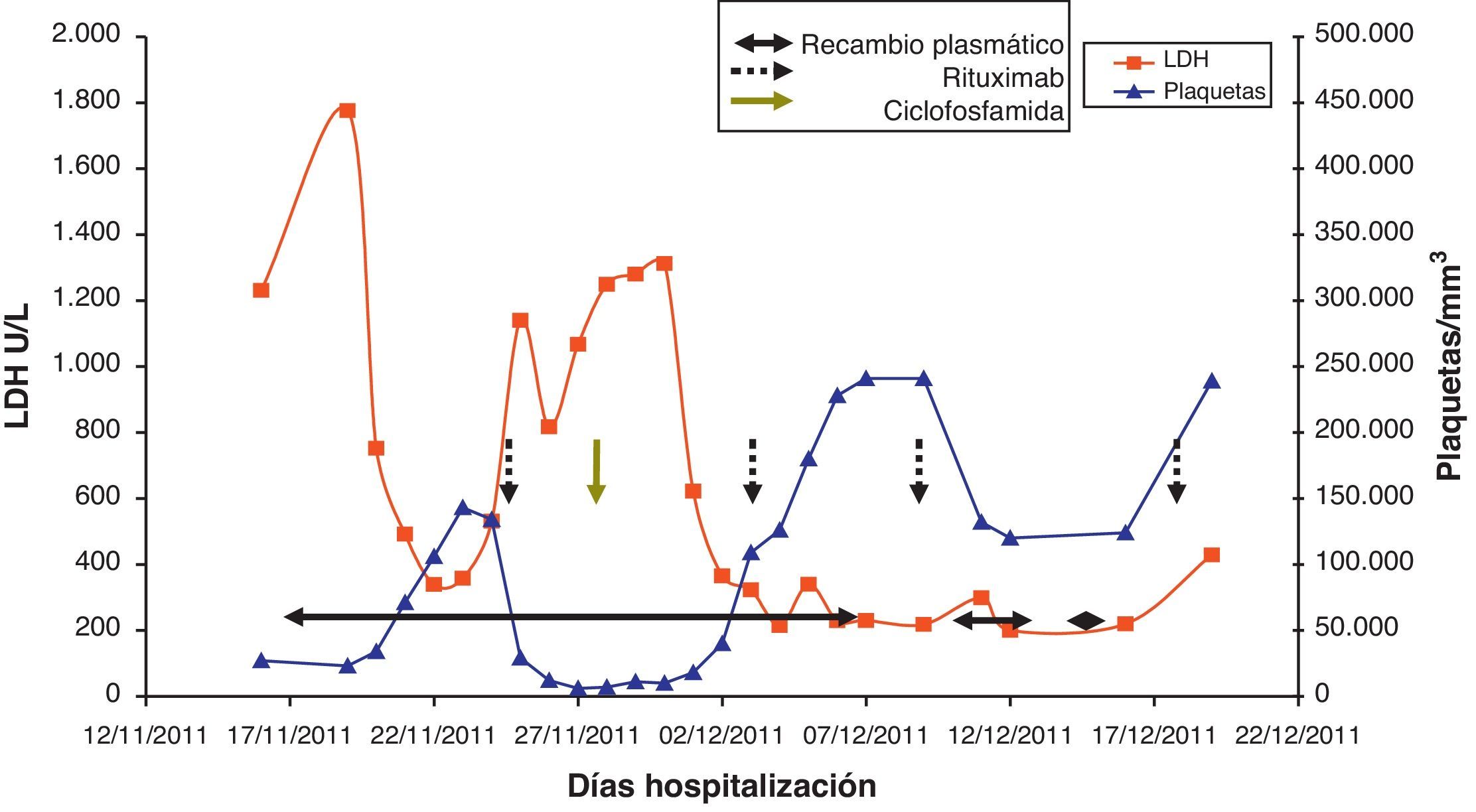
Caso clínicoMujer de 47 años de edad, que acude a urgencias en diciembre del 2011 por epigastralgia inespecífica de varios días de evolución. Se trata de una paciente diagnosticada en 1998 de LES con artritis palindrómica, úlceras orales, serología anti-ADN positiva y anticuerpos antinucleares positivos, y pleuropericarditis con derrame pleural izquierdo en 2001. En los últimos 4 años no manifiesta clínica lúpica, manteniendo tratamiento con prednisona 10mg/d, hidroxicloroquina 200mg/12h y omeprazol. En urgencias, la exploración clínica revela buen estado general, sin alteraciones a nivel neurológico, cardíaco ni pulmonar, apirética y normotensa, con algunas equimosis en el abdomen y los brazos. La analítica de sangre a su ingreso destacaba: hemoglobina 10,5g/dl, 27.000 plaquetas/mm3, LDH 1.231 U/l, gammaglutamil transpeptidasa 70 U/L, transaminasa glutámico pirúvica 54 U/L, bilirrubina total 1,40mg/dl y proteína C reactiva 32mg/l, con urea y creatinina normales. Las exploraciones complementarias, electrocardiograma, radiografía de tórax y abdomen y ecografía abdominal, fueron normales. Las alteraciones hematológicas y bioquímicas sugerían una hemólisis y trombopenia de probable origen inmunitario, motivo por el que ingresó para estudio, aumentando la dosis habitual de corticoides a 0,5mg/kg de prednisona. Durante los primeros días de ingreso, la trombopenia empeora hasta 17.000 plaquetas/mm3 y a los 4 días la paciente presenta clínica neurológica brusca con alteración del lenguaje y pérdida de fuerza en la mano derecha, que se recupera totalmente en pocas horas. La resonancia magnética vascular cerebral revela múltiples infartos lacunares agudos en el hemisferio cerebeloso derecho y frontal izquierdo. En analíticas posteriores, el proteinograma, la función tiroidea, los parámetros renales y la bioquímica de orina fueron normales. Las determinaciones de anticoagulante lúpico y test de Coombs directo fueron negativas, con anticuerpos antinucleares 1/160 (normal < 1/80) y haptoglobina < 10. El Servicio de Hematología confirmó anemia Coombs negativa, de características hemolíticas (aumento de LDH, bilirrubina indirecta y reticulocitos con disminución de haptoglobina). Se revisó la morfología de sangre periférica, detectándose un aumento de esquistocitos con presencia de punteado basófilo eritrocitario, y cuerpos de Howell-Jolly (fig. 1), confirmando anemia hemolítica microangiopática. Junto con la trombopenia y las alteraciones neurológicas, el diagnóstico clínico final fue de PTT. Inmediatamente, se inició corticoterapia a dosis altas (1,5mg metilprednisolona por kg de peso al día) y recambios plasmáticos (40ml/kg del paciente cada día), suspendiéndose el tratamiento con hidroxicloroquina. Tras 3 sesiones de recambios plasmáticos, los recuentos plaquetarios y los niveles de LDH se normalizaron, pero 2 días después dichos parámetros se deterioraron de nuevo, alcanzándose una cifra de plaquetas < 10.000/mm3. Dada la refractariedad al tratamiento, se decide añadir una segunda línea terapéutica con RTX en pauta de 375mg/m2 cada semana durante 4 semanas. Tras la primera dosis de RTX, se planeó suspender los recambios plasmáticos durante 48 h para incrementar su eficacia, pero tras 24 h sin plasmaféresis la paciente muestra nuevo descenso de hemoglobina (Hb: 5,6g/dl) incremento de LDH y de esquistocitos en sangre periférica, así como nueva sintomatología neurológica consistente en estado confusional, afasia motora y debilidad. Se decide reiniciar de nuevo de forma urgente los recambios plasmáticos diarios y se administra un pulso de ciclofosfamida i.v. (750mg/m2). La sintomatología neurológica se recupera rápidamente y después de 2 días los niveles de plaquetas y de LDH vuelven a mejorar hasta normalizarse. Tras 4 días con respuesta completa, se empiezan a espaciar los recambios plasmáticos hasta suspenderse tras un total de 21 sesiones, al tiempo que se reduce la pauta de esteroides hasta 10mg/d de prednisona. A los 4 meses del alta se mantiene la remisión hematológica completa, sin requerir otro tratamiento que prednisona a dosis bajas. Los niveles de ADAMTS13 obtenidos en el momento inicial eran de 0% y los extraídos a los 2 meses después de la finalización del tratamiento se mantienen igual. En la figura 2 se observa el desarrollo del proceso patológico y los tratamientos utilizados.
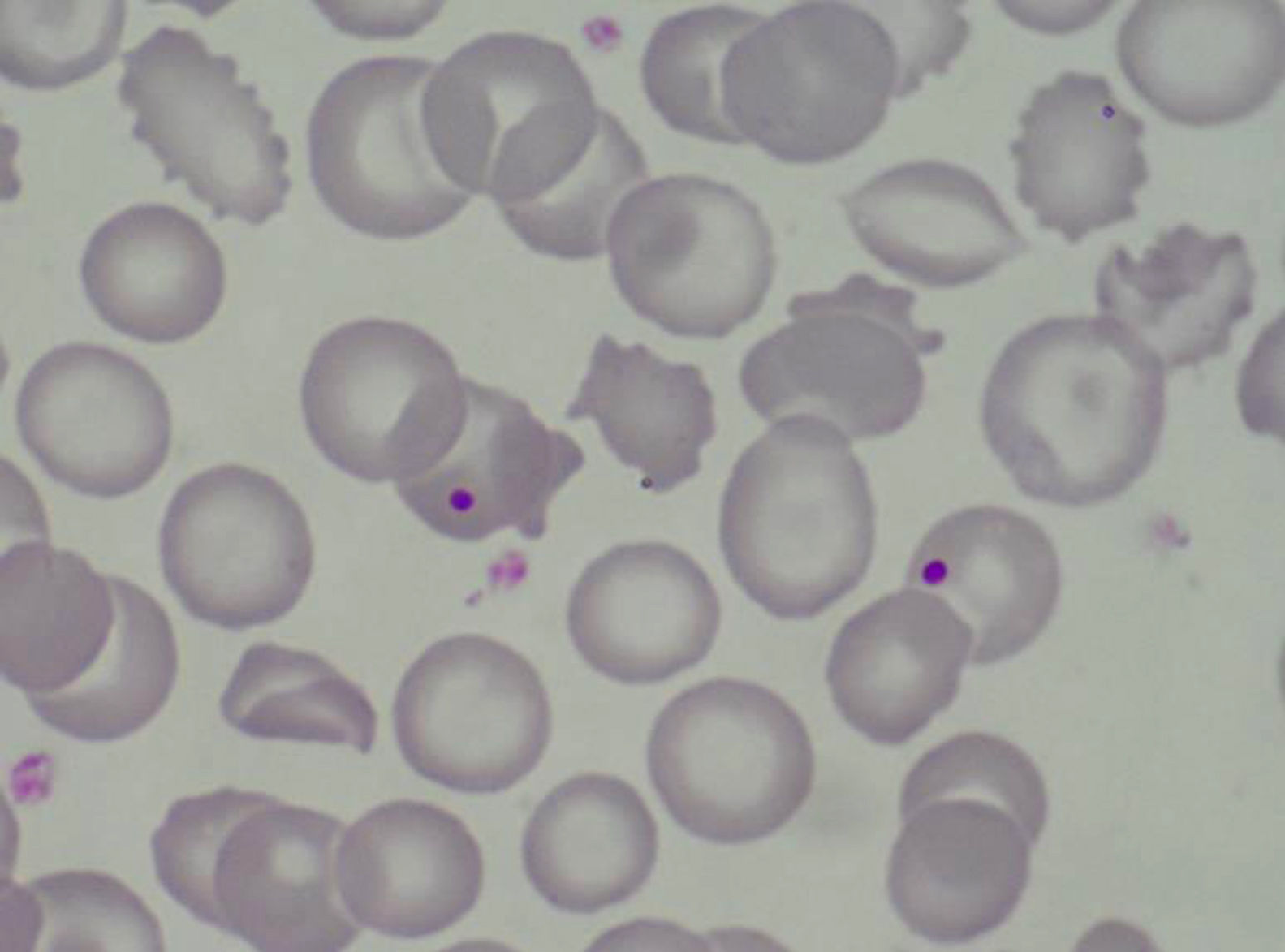
La PTT se caracteriza por anemia hemolítica microangiopática, trombocitopenia, manifestaciones neurológicas fluctuantes y trastornos renales. La oclusión de arteriolas y capilares por microtrombos compuestos fundamentalmente por plaquetas es típica de este trastorno y consecuencia de la mayor agregación plaquetar asociada a la presencia de grandes multímeros de factor von Willebrand (Fv W), presumiblemente debido a la disminución de la actividad de la enzima ADAMTS13, encargada de escindir estos multímeros2,4,5. Se ha sugerido que la patogenia de la PTT en el LES podría estar relacionada con la inhibición de la acción de la metaloproteinasa ADAMTS13 por autoanticuerpos10 pero no hay acuerdo unánime en este sentido.
Aunque la asociación de LES y PTT está descrita en la literatura, no es una asociación frecuente y es difícil su diagnóstico. Recientemente, se ha publicado una serie de 283 pacientes con PTT recopilados a lo largo de 20 años6, incluyendo determinaciones de niveles de ADAMTS13. Se consideró nivel bajo si había < 10% de la actividad normal de ADAMTS13. En el grupo estudiado, 107 casos eran idiopáticos (37,8%), 21 casos (7,4%) secundarios a lupus y 16 casos (5,6%) secundarios a otros procesos inmunitarios. El nivel de ADAMTS13 fue bajo en casi la mitad de los casos idiopáticos (48%), pero solo 2 casos con LES (10%) y uno con otras enfermedades inmunitarias (7%) lo tuvieron reducido. Cuando los autores revisan las asociaciones clínicas de los casos idiopáticos comprueban cómo aquellos con los niveles bajos de ADAMTS13 eran enfermos más jóvenes (41 vs. 57 años), tenían menor número de plaquetas al diagnóstico (17.000/mm3 vs. 47.000/mm3), menos afectación renal, necesidad de más sesiones de recambios plasmáticos (19 vs. 9) y más posibilidad de recurrencia (40% vs. 7%). En otro trabajo, Letchumanan et al.9 estudiaron 10 casos de PTT idiopática y 8 casos de PTT asociada a lupus. Típicamente, los casos de PTT asociados a lupus ocurrían en fase de actividad de la enfermedad y tenían más compromiso renal. Estos datos indican que el caso aquí presentado tiene un perfil más similar a la PTT idiopática con supresión de ADAMTS136. Coinciden el perfil de edad, la trombopenia más acusada, la ausencia de afectación renal, el mayor número de recambios plasmáticos necesarios y la refractariedad. Sin embargo, el inicio del proceso tuvo un comportamiento atípico, ya que no presentó actividad lúpica ni afectación renal en el momento del diagnóstico9. En otros aspectos, nuestro caso mostró 2 de los principales problemas que ocasiona la asociación entre LES y PTT. El primero de ellos es la dificultad del diagnóstico, ya que la presentación clínica puede confundirse con signos propios de LES: anemia hemolítica, trombopenia y dolor abdominal. La revisión del frotis sanguíneo y la detección de esquistocitos es imprescindible para categorizar la anemia hemolítica como microangiopática. La ausencia de positividad al test de Coombs, junto al resto de los síntomas, con trombopenia y alteración neurológica, apoyan el diagnóstico de PTT y obligan a un inicio de recambios plasmáticos de forma urgente. Los niveles bajos de ADAMTS13 corroboran aún más el diagnóstico de PTT. Letchumannan et al., en su trabajo de comparación entre casos de PTT idiopáticos y los asociados a lupus, informaron que se tarda casi 2 semanas más en diagnosticar los casos asociados a LES que los casos idiopáticos (19,5 vs. 7,9 días)9. El segundo problema es la peor respuesta al tratamiento. En el mismo estudio citado, se encontró que el tiempo de respuesta fue más lento en los casos secundarios a LES que en los idiopáticos (31,3 días vs. 16,8), que fue necesario el uso de mayores dosis de citotóxicos, inmunosupresores y agentes biológicos, y que la mortalidad fue más alta en el grupo con LES9. En nuestra paciente fracasó la primera línea terapéutica, siendo preciso añadir al esquema inicial de recambios plasmáticos y corticoides un tratamiento más agresivo con agentes inmunosupresores que han demostrado buenos resultados en estos casos, como RTX5-8,11,12 y ciclofosfamida13.
En resumen, aportamos una paciente con una asociación poco frecuente, LES y PTT, asociada a una disminución de la actividad de ADAMTS13. El caso es destacable porque la enferma no tenía actividad lúpica como suele ser habitual y llamamos la atención de la dificultad de diagnóstico inicial y del comportamiento agresivo y refractario que precisó tratamiento inmunosupresor.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


