


Lymphadenopathy syndrome is a common manifestation of systemic lupus erythematosus (SLE). In general, enlarged lymph nodes are small and can be found in the cervical, inguinal and axillary regions. Lymph node involvement is present in up to 25% of the patients and normally appears in the first stages of the disease or during relapses.1 The differential diagnosis of lymphadenopathy syndrome in SLE includes histiocytic necrotizing lymphadenitis or Kikuchi-Fujimoto disease (KFD), Castleman's disease, syphilis, tuberculosis, sarcoidosis, infectious mononucleosis (Epstein–Barr virus [EBV], cytomegalovirus), herpes simplex, human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), other infections and lymphoma.2
Kikuchi-Fujimoto disease, or histiocytic necrotizing lymphadenitis, is a rare clinical disorder characterized by lymph node involvement (generally cervical), fever, night sweats and leukopenia. It mainly affects young Asian women. It was first described by Kikuchi and Fujimoto in 1972.3,4 The associated mortality is increased by heart, lung and liver involvement. The exact cause of KFD is not yet known, but recent publications are inclined toward a viral infection (EBV and others) or an autoimmune disorder (an exaggerated immune response mediated by T cells).5 To diagnosis the disease, it is necessary to perform an excisional biopsy of the affected lymph nodes. The association between KFD and SLE is rare, and whether it is incidental or a clinical manifestation of SLE continues to be a matter of debate.
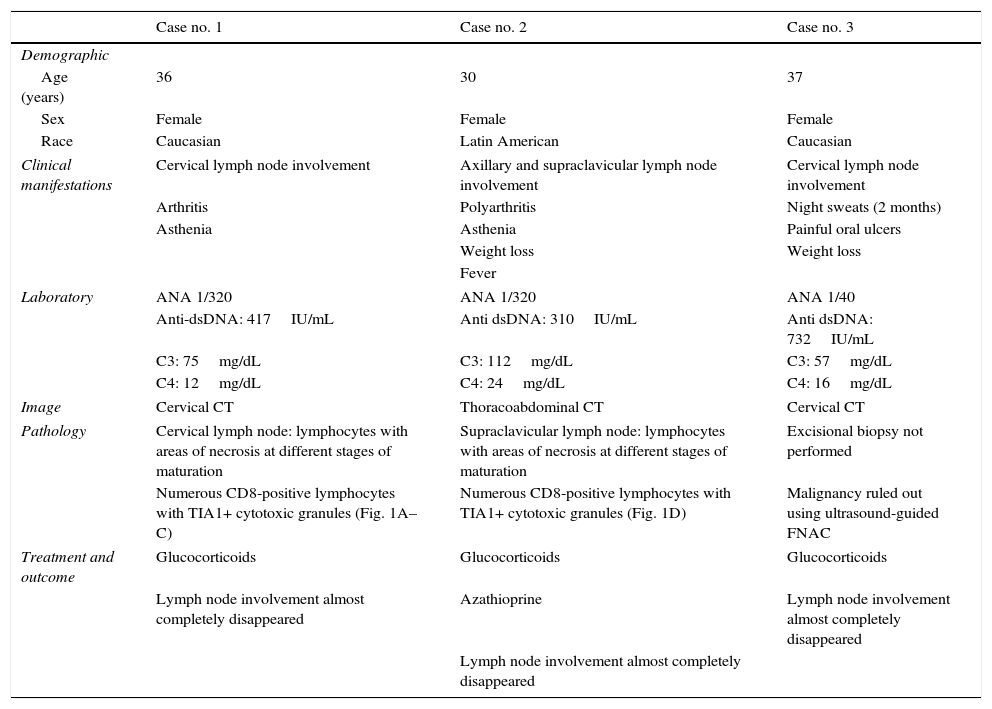
The coexistence of these 2 diseases is reported in 2 young SLE patients (cases nos. 1 and 2) and we describe another case of lymphadenopathy syndrome in SLE (case no. 3) in which the affected lymph node was studied by cytology rather than excisional biopsy (Table 1). In all 3 cases, the results of serological testing for HIV, HBV, HCV, EBV, cytomegalovirus, herpes simplex virus, rubella, toxoplasma, parvovirus B19, Yersinia enterocolitica, Salmonella and Brucella were negative, as were the results of interferon-γ release assays (IGRA) for all 3 patients.
Description of the Three Cases.
| Case no. 1 | Case no. 2 | Case no. 3 | |
|---|---|---|---|
| Demographic | |||
| Age (years) | 36 | 30 | 37 |
| Sex | Female | Female | Female |
| Race | Caucasian | Latin American | Caucasian |
| Clinical manifestations | Cervical lymph node involvement | Axillary and supraclavicular lymph node involvement | Cervical lymph node involvement |
| Arthritis | Polyarthritis | Night sweats (2 months) | |
| Asthenia | Asthenia | Painful oral ulcers | |
| Weight loss | Weight loss | ||
| Fever | |||
| Laboratory | ANA 1/320 | ANA 1/320 | ANA 1/40 |
| Anti-dsDNA: 417IU/mL | Anti dsDNA: 310IU/mL | Anti dsDNA: 732IU/mL | |
| C3: 75mg/dL | C3: 112mg/dL | C3: 57mg/dL | |
| C4: 12mg/dL | C4: 24mg/dL | C4: 16mg/dL | |
| Image | Cervical CT | Thoracoabdominal CT | Cervical CT |
| Pathology | Cervical lymph node: lymphocytes with areas of necrosis at different stages of maturation | Supraclavicular lymph node: lymphocytes with areas of necrosis at different stages of maturation | Excisional biopsy not performed |
| Numerous CD8-positive lymphocytes with TIA1+ cytotoxic granules (Fig. 1A–C) | Numerous CD8-positive lymphocytes with TIA1+ cytotoxic granules (Fig. 1D) | Malignancy ruled out using ultrasound-guided FNAC | |
| Treatment and outcome | Glucocorticoids | Glucocorticoids | Glucocorticoids |
| Lymph node involvement almost completely disappeared | Azathioprine | Lymph node involvement almost completely disappeared | |
| Lymph node involvement almost completely disappeared | |||
ANA, antinuclear antibodies; CT, computed tomography; dsDNA, double-stranded DNA; FNAC, fine-needle aspiration cytology.
Kikuchi-Fujimoto disease has been associated with SLE and other connective tissue diseases, such as antiphospholipid syndrome, Sjögren's syndrome, relapsing polychondritis and autoimmune hepatitis.6,7 It is generally a benign disease that resolves spontaneously in 1–4 months, but cases with a poor outcome and atypical presentation have been reported.8 Biopsy of the affected lymph nodes reveals irregular necrotized regions, mostly in the paracortical areas, with partial or complete loss of the follicular architecture, karyorrhexis and the absence of neutrophils, with the presence of transformed lymphocytes (immunoblasts) around the necrotic areas. The lesional cells have also been reported to infiltrate the perinodal fibroadipose tissue.9 Findings that are not common in KFD are skin lesions, mesenteric lymph node involvement, splenomegaly, myalgia and parotid swelling.
There is no effective treatment but, in cases without complications or in self-limiting disease, glucocorticoids are usually efficacious, as in the 3 cases documented here, although studies reveal that the rate of recurrence of the disease is 3%–4%.10
In the third case, excisional biopsy was not performed, a fact that means that, in this patient, we cannot speak of histiocytic necrotizing lymphadenitis. However, the course and outcome of the disease are consist with that possibility (the good response to glucocorticoids rules out an infectious or malignant disease). The early diagnosis is fundamental for the correct management of the disease, and the association with SLE requires more research. Our experience supports the hypothesis that it is necessary to perform excisional biopsy of the lymph nodes in cases of lymphadenopathy syndrome in SLE that have not been diagnosed. Thus, not only is it necessary to exclude malignancy by means of cytology; the best alternative is to completely remove the lymph node to attempt to identify the disorder, because the therapeutic approach will be different in each case depending on the result (Fig. 1).
 Hematoxylin-eosin: cervical lymph node. Lymphocytes with areas of necrosis at different stages of maturation. (B) Cervical lymph node. Presence of lymphocytes expressing CD8 in relation to areas of necrosis. (C) Cervical lymph node. Presence of lymphocytes expressing TIA1 in relation to the areas of necrosis. (D) Supraclavicular lymph node. Hematoxylin-eosin: lymphocytes with areas of necrosis at different stages of maturation.")
(A) Hematoxylin-eosin: cervical lymph node. Lymphocytes with areas of necrosis at different stages of maturation. (B) Cervical lymph node. Presence of lymphocytes expressing CD8 in relation to areas of necrosis. (C) Cervical lymph node. Presence of lymphocytes expressing TIA1 in relation to the areas of necrosis. (D) Supraclavicular lymph node. Hematoxylin-eosin: lymphocytes with areas of necrosis at different stages of maturation.
The authors declare they have no conflicts of interest.
Please cite this article as: Salman-Monte TC, Pérez Ruiz J, Almirall M, Campillo Ibáñez MÁ, Barranco Sanz LC, Carbonell Abelló J. Síndrome poliadenopático en lupus eritematoso sistémico: ¿es la enfermedad de Kikuchi-Fujimoto? Reumatol Clin. 2017;13:55–56.


