


El síndrome poliadenopático es una manifestación frecuente en el lupus eritematoso sistémico (LES). Por lo general, las adenopatías son de pequeño tamaño y se encuentran en la región cervical, inguinal y axilar. Están presentes en hasta el 25% de los pacientes y normalmente aparece en las primeras etapas de la enfermedad o en las recaídas1. El diagnóstico diferencial del síndrome poliadenopático en el LES incluye la linfadenitis histiocítica necrosante o enfermedad de Kikuchi-Fujimoto (EKF), la enfermedad de Castleman, la sífilis, la tuberculosis, la sarcoidosis, los síndromes mononucleósidos (virus de Epstein-Barr [EBV], citomegalovirus), el herpes simple, el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB) y hepatitis C (VHC), otras infecciones y linfoma2.
La EKF o linfadenitis histiocítica necrosante es un raro trastorno clínico caracterizado por adenopatías (generalmente cervicales), fiebre, sudores nocturnos y leucopenia. Afecta principalmente a mujeres jóvenes asiáticas. Fue descrita por primera vez por Kikuchi y Fujimoto en 19723,4. Las afectaciones cardíaca, pulmonar y hepática aumentan la mortalidad en dicha entidad. La causa exacta de la EKF es aún desconocida, pero las publicaciones recientes se inclinan hacia una infección viral (EBV y otros) o trastorno autoinmune (respuesta inmunitaria exagerada mediada por células T)5. Para diagnosticar la enfermedad es necesario realizar una biopsia escisional de los ganglios linfáticos afectados. La asociación de la EKF con el LES es rara y sigue siendo un tema de debate si se trata de una coincidencia casual o una manifestación clínica en el LES.
Se presenta la coexistencia de estas 2 enfermedades en 2 jóvenes pacientes con LES (casos 1 y 2) y describimos otro caso de síndrome poliadenopático en el LES (caso 3) en el que se realizó citología del ganglio linfático afectado en lugar de biopsia escisional (tabla 1). En los 3 casos los resultados de las serologías de VIH, VHB, VHC, EBV, citomegalovirus, virus del herpes simple, rubéola, toxoplasma, parvovirus B19, Yersinia enterocolitica, Salmonella y Brucella fueron negativos, al igual que el resultado de los ensayos de liberación de interferón γ (IGRA, por sus siglas en inglés) para las 3 pacientes.
Descripción de los 3 casos
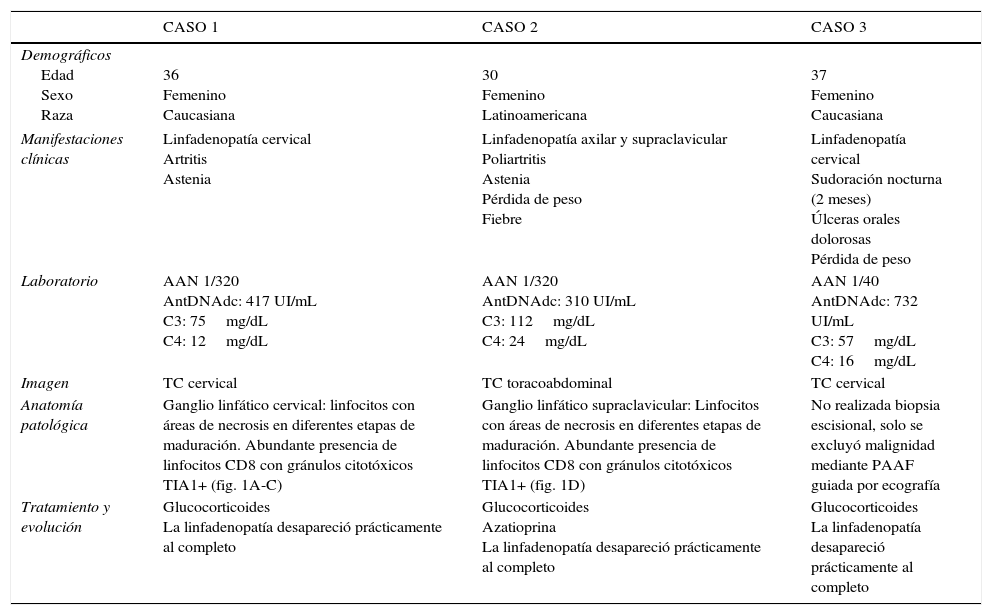
| CASO 1 | CASO 2 | CASO 3 | |
|---|---|---|---|
| Demográficos Edad Sexo Raza | 36 Femenino Caucasiana | 30 Femenino Latinoamericana | 37 Femenino Caucasiana |
| Manifestaciones clínicas | Linfadenopatía cervical Artritis Astenia | Linfadenopatía axilar y supraclavicular Poliartritis Astenia Pérdida de peso Fiebre | Linfadenopatía cervical Sudoración nocturna (2 meses) Úlceras orales dolorosas Pérdida de peso |
| Laboratorio | AAN 1/320 AntDNAdc: 417 UI/mL C3: 75mg/dL C4: 12mg/dL | AAN 1/320 AntDNAdc: 310 UI/mL C3: 112mg/dL C4: 24mg/dL | AAN 1/40 AntDNAdc: 732 UI/mL C3: 57mg/dL C4: 16mg/dL |
| Imagen | TC cervical | TC toracoabdominal | TC cervical |
| Anatomía patológica | Ganglio linfático cervical: linfocitos con áreas de necrosis en diferentes etapas de maduración. Abundante presencia de linfocitos CD8 con gránulos citotóxicos TIA1+ (fig. 1A-C) | Ganglio linfático supraclavicular: Linfocitos con áreas de necrosis en diferentes etapas de maduración. Abundante presencia de linfocitos CD8 con gránulos citotóxicos TIA1+ (fig. 1D) | No realizada biopsia escisional, solo se excluyó malignidad mediante PAAF guiada por ecografía |
| Tratamiento y evolución | Glucocorticoides La linfadenopatía desapareció prácticamente al completo | Glucocorticoides Azatioprina La linfadenopatía desapareció prácticamente al completo | Glucocorticoides La linfadenopatía desapareció prácticamente al completo |
La EKF se ha descrito en asociación con el LES y en otras enfermedades del tejido conectivo, tales como el síndrome antifosfolipídico, el síndrome de Sjögren, la policondritis recidivante y la hepatitis autoinmune6,7. Por lo general, es una enfermedad con curso benigno y generalmente se resuelve espontáneamente en uno a 4 meses, pero algunos casos han sido reportados con un mal resultado y presentaciones atípicas8. La biopsia de los ganglios linfáticos afectados muestra las regiones necrosantes irregulares principalmente en las áreas paracorticales con pérdida parcial o completa de la arquitectura folicular, cariorrexis y ausencia de neutrófilos, con presencia de linfocitos transformados (inmunoblastos) alrededor de las áreas necróticas. Se ha documentado además que las células lesionales infiltran el tejido fibroadiposo perinodal9. Manifestaciones poco frecuentes de la EKF son: lesiones cutáneas, adenopatías mesentéricas, esplenomegalia, mialgias y parotidomegalia.
No existe un tratamiento eficaz, pero en los casos sin complicaciones o enfermedad autolimitada el tratamiento con glucocorticoides suele ser eficaz, como en los 3 casos documentados, aunque los estudios revelan la recurrencia de la enfermedad en el 3-4% de los pacientes10.
En el tercer caso no se realizó biopsia escisional, hecho que evita que podamos informar de otro caso de linfadenitis histiocítica necrosante, pero el curso y la evolución de la enfermedad baraja esta posibilidad (la buena respuesta a los glucocorticoides descarta origen infeccioso o malignidad en el cuadro). El diagnóstico precoz es fundamental para la correcta gestión de la enfermedad y la asociación con el LES necesita más investigación. Nuestra experiencia refuerza la hipótesis de que es necesario realizar biopsia escisional de los ganglios linfáticos en los casos de síndrome poliadenopático en el LES que carezcan de diagnóstico. Por lo tanto, no solo hay que descartar malignidad mediante citología, es más aconsejable extraer el ganglio completo para intentar filiar el cuadro porque la actitud terapéutica será diferente en cada caso dependiendo del resultado (fig. 1).
 Hematoxilina-eosina: ganglio linfático cervical. Linfocitos con áreas de necrosis en diferentes etapas de maduración. B) Ganglio linfático cervical. Presencia de linfocitos con expresión CD8 en relación con áreas de necrosis. C) Ganglio linfático cervical. Presencia de linfocitos con expresión TIA1 en relación con áreas de necrosis. D) Ganglio linfático supraclavicular. Hematoxilina-eosina: linfocitos con áreas de necrosis en diferentes etapas de maduración.")
A) Hematoxilina-eosina: ganglio linfático cervical. Linfocitos con áreas de necrosis en diferentes etapas de maduración. B) Ganglio linfático cervical. Presencia de linfocitos con expresión CD8 en relación con áreas de necrosis. C) Ganglio linfático cervical. Presencia de linfocitos con expresión TIA1 en relación con áreas de necrosis. D) Ganglio linfático supraclavicular. Hematoxilina-eosina: linfocitos con áreas de necrosis en diferentes etapas de maduración.
Los autores declaran no tener conflicto de intereses.


