








To describe the clinical and laboratory data, with special emphasis on thoracic imaging findings, in 14 patients with a definitive diagnosis of granulomatosis with polyangiitis (GPA).
MethodsThe clinical and tomographic data of 14patients with a definitive diagnosis of GPA are presented. Patients with thoracic manifestations suggestive of GPA were evaluated in 3 hospitals from 2000 to 2012. All patients had a sputum analysis and bronchoalveolar lavage for bacterial, mycobacterial and fungal stains and cultures; antineutrophil cytoplasmic antibodies, antinuclear-antibodies, rheumatoid factor, and a biopsy of involved organs.
ResultsA total of 13 patients had at least two organs involved. The most frequent thoracic findings were pulmonary nodules, ground glass opacities and patches of consolidation; other abnormalities were tracheal stenosis, diffuse alveolar hemorrhage, lung masses with organized pneumonia. More than three-quarters (78%) of patients had positive antineutrophil cytoplasmic antibodies (ANCA). Ten patients had respiratory tissue biopsy (8 open lung, one tracheal, and one nasal). In 4 patients the diagnosis was made with the classic organ involvement in GPA, positive ANCA, and renal or skin biopsy, and response to treatment on follow-up. At 6–12months all patients showed clinical and radiological improvement, with 54% showing a recurrence of disease.
DiscussionThe majority of thoracic findings described in GPA are presented in this study. A complete diagnostic approach with invasive diagnostic procedures to rule out other more prevalent respiratory diseases with similar thoracic manifestations must be performed. The positivity of ANCA in this study was high, and the recurrence of the disease was frequent.
Describir las diversas manifestaciones clínicas, de laboratorio e imagenológicas torácicas en 14 pacientes con poliangeítis granulomatosa (Wegener) (PAG).
Material y métodosPresentamos los datos clínicos, de laboratorio e imagen de pacientes con diagnóstico de PAG. Se incluyeron pacientes con manifestaciones torácicas sugerentes de PAG evaluados en 3 hospitales entre los años 2000 y 2012. A todos los pacientes les realizamos tinciones y cultivos bacteriológicos, para micobacterias y hongos, en expectoración y lavado broncoalveolar; anticuerpos anticitoplasma de neutrófilos (ANCA), anticuerpos antinucleares, factor reumatoide y biopsia de órganos afectados.
ResultadosTrece pacientes tuvieron afección de por lo menos 2 órganos. Las manifestaciones torácicas fueron nódulos pulmonares, opacidades en vidrio esmerilado y parches de consolidación; otras anormalidades fueron estenosis de tráquea, hemorragia alveolar difusa, masas pulmonares con neumonía organizada. El 78% tuvo ANCA positivos (la mayoría c-ANCA). A 10 pacientes se les realizó biopsia de tejido respiratorio (8 de pulmón, una de tráquea y una nasal). En 4 pacientes el diagnóstico se sustentó con la afección de diversos órganos, ANCA positivos, biopsia de riñón o piel y respuesta a tratamiento. A los 6-12 meses todos los pacientes mostraron mejoría clínica e imagenológica, y el 54% de 11 pacientes en seguimiento presentaron recaída.
ConclusionesEn esta serie de casos presentamos la mayoría de las manifestaciones radiológicas torácicas descritas en pacientes con PAG. Enfatizamos el realizar un abordaje diagnóstico ordenado para descartar otras patologías con prevalencia más alta que se expresan con manifestaciones pulmonares similares. La frecuencia de ANCA positivos fue elevada y la recurrencia de enfermedad, frecuente.
Granulomatous polyangiitis (GPA) is the recently accepted name for Wegener's granulomatosis1 and is defined as a systemic disease with necrotizing vasculitis that affects vessels of small and medium caliber (capillaries, arterioles, venules, arteries) with granulomatous inflammation, affecting the respiratory tract (upper and lower), and frequently the kidneys, with glomerulonephritis and focal necrosis.2–5 It has a prevalence of 25–160 cases per million inhabitants and an incidence of 9–16 cases per million per year.6,7 Respiratory involvement is usually the most prevalent sign at the onset of the disease and occurs in up to 90% of cases during its evolution and is characteristically caused by granulomatous necrotizing inflammation, and its associated complications, and/or capillaritis.2–5 Frequently chest X-ray and tomographic manifestations are pulmonary nodules, which in most patients are bilateral and, in 50%, accompanied by cavitation.8–13 Opacities of various types are presented, from low-density diffuse ground glass images, to low-density patches of consolidations with poorly defined borders with or without cavitation8–13 in half of the patients. In 15%–30% of cases there is tracheobronchial involvement. The objective of this paper is to present the clinical manifestations, laboratory findings and the various imaging abnormalities found in a cohort of 14 patients with GPA during the initial presentation and after treatment.
MethodsThose included were hospitalized patients presenting with respiratory disease in 3 hospitals between 2000 and 2012 who met the Chapell Hill 2 diagnostic criteria for GPA, in the city of San Luis Potosi (Mexico). San Luis Potosi is a state with 2585518 inhabitants and receives patients referred from other states of the country, including 10 neighbors. Clinical, laboratory and images were collected retrospectively. All patients were evaluated jointly by rheumatologists and respiratory medicine specialists.
All patients underwent blood count, blood chemistry, C-reactive protein, liver function tests, determination of anti-neutrophil cytoplasmic antibodies (ANCA) by immunofluorescence, antinuclear antibodies, rheumatoid factor and computed tomography (CT) of the chest; 12 and creatinine clearance was determined. 2 patients with diffuse alveolar hemorrhage underwent anti-glomerular basement membrane antibody determination. All patients had samples of sputum and bronchoalveolar lavage, which included Gram staining, Ziehl-Nilssen, PAS and Grocott, and cultures for bacteria, mycobacteria and fungi. In 5 of the 10 patients with scattered pulmonary nodules, transthoracic echocardiography and blood cultures were performed. In 10 patients respiratory tissue biopsies (8 lung, one tracheal and one nasal) were obtained; 2 underwent a renal biopsy and 3 a skin biopsy.
Eleven patients were followed by the outpatient clinic after hospitalization and underwent chest CT 6–12 months after starting treatment.
ResultsEleven patients (84%) were male with a mean age of 44 years. The average duration of symptoms was 5.3 months. Clinical manifestations are described in Table 1. In 10 of the 12 patients with creatinine clearance, the determination of GFR was normal (values 100–120ml/min), one presented 80ml/min and one 10ml/min. All patients who underwent bronchoalveolar lavage were negative for any type of germ. In the 2 cases with diffuse alveolar hemorrhage and antinuclear antibodies, anti-glomerular basement membrane antibodies were negative and bronchoalveolar lavage was diagnostic of the alveolar hemorrhage.
Symptoms and Systemic Manifestations in 14 Patients With Granulomatous Polyangiitis.
| Cases, n (%) | Cases, n (%) | ||
| Mean age | 44 years | Sinusitis | 9 (64) |
| Male | 11 (78) | Arthralgia/arthritis | 9 (64) |
| Involvement of 2 or more organs | 13 (92) | Cutaneous vasculitis | 4 (28) |
| Fever | 7 (54) | Erythrocyturia | 4 (28) |
| Cough | 9 (64) | Decreased renal function | 2 (14) |
| Dyspnea | 7 (50) | Episcleritis/uveitis | 3 (23) |
| Hemoptysis | 3 (23) | Mononeuritis multiplex | 4 (28) |
| Nasal obstruction | 9 (64) | ANCA positive | 11 (78) |
ANCA: antineutrophil cytoplasmic antibodies.
Final diagnosis was obtained through a biopsy of airway tissue in 10 patients (71%): 8, lung tissue, one trachea and one nasal; 3 were ANCA positive (Table 2), with all biopsies demonstrating granulomas with necrosis and vasculitis as the main finding. In the 4 (29%) cases in whom respiratory tissue biopsies were not performed the diagnosis was established with the combination of: evidence of systemic disease, pulmonary nodules, sinusitis, microhematuria, impaired renal function, arthralgia or arthritis, cutaneous vasculitis, mononeuritis multiple, positive ANCA; in addition, 2 of these cases had a skin biopsy performed and in the other 2, a kidney biopsy, which showed leukocytoclastic vasculitis without antibodies and pauci-immune glomerulonephritis, focal or segmental necrosis, respectively (Table 2). ANCA were positive in 11 patients (78%), 8 cytoplasmic and 3 perinuclear pattern. Only one patient of the 3 presented negative ANCA, with a disease limited to cavitary pneumonia and sinusitis.
Systemic Manifestations, ANCA Results and Biopsy Site in 14 Patients With Granulomatous Polyangiitis.
| Age, gender | Respiratory manifestation | Locomotor | Renal | Eye/Ear | Cutaneous | Neurological | ANCA | Biopsy site |
| 30, F | Cavitated consolidation | Joint pain | – | – | – | – | Negative | Lung |
| 20, F | Tracheal stenosis | Joint pain | – | – | Giant ulcer | – | Negative | Trachea |
| 53, M | PN | Arthritis | Erythrocyturia | – | – | MM | positive c-ANCA | Lung |
| 52, M | Cavitated consolidation | Joint pain | – | Episcleritis | – | – | p-ANCA positive | Lung |
| 34, M | Diffuse alveolar hemorrhage, sinusitis | Joint pain | Chronic renal failure | – | Vasculitis skin | MM | positive c-ANCA | Skin |
| 53, M | Diffuse alveolar hemorrhage, sinusitis and PN | – | Focal segmental glomerulonephritis | – | Cutaneous vasculitis | – | c-ANCA positive, negative AAN | Renal |
| 60, F | Masses and PN | Joint pain | – | Hearing loss | – | – | p-ANCA positive | Lung |
| 55, M | PN with and without cavitation, sinusitis | Arthritis | Decreased renal function | – | – | – | positive c-ANCA | Renal |
| 46, M | Pulmonary nodules | – | Erythrocyturia | – | – | – | Negative | Lung |
| 42, M | Pulmonary nodules | – | – | – | MM | positive c-ANCA | Lung | |
| 43, M | PN and sinusitis | Erythrocyturia | – | – | – | positive c-ANCA | Lung | |
| 52, M | PN and sinusitis | – | – | Episcleritis | – | MM | p-ANCA positive | Lung |
| 38, M | PN and sinusitis | Arthritis | – | Uveitis | – | – | positive c-ANCA | Nasal |
| 48, M | PN and sinusitis | Arthritis | Erythrocyturia | – | Skin vasculitis | – | positive c-ANCA | Skin |
ANA: antinuclear antibodies; ANCA: antineutrophil cytoplasmic antibodies; c-ANCA: cytoplasmic antineutrophil cytoplasmic antibodies; F: female, M: male; MM: mononeuritis multiplex; PN: pulmonary nodules; p-ANCA: perinuclear antineutrophil cytoplasmic antibodies.
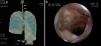
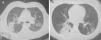
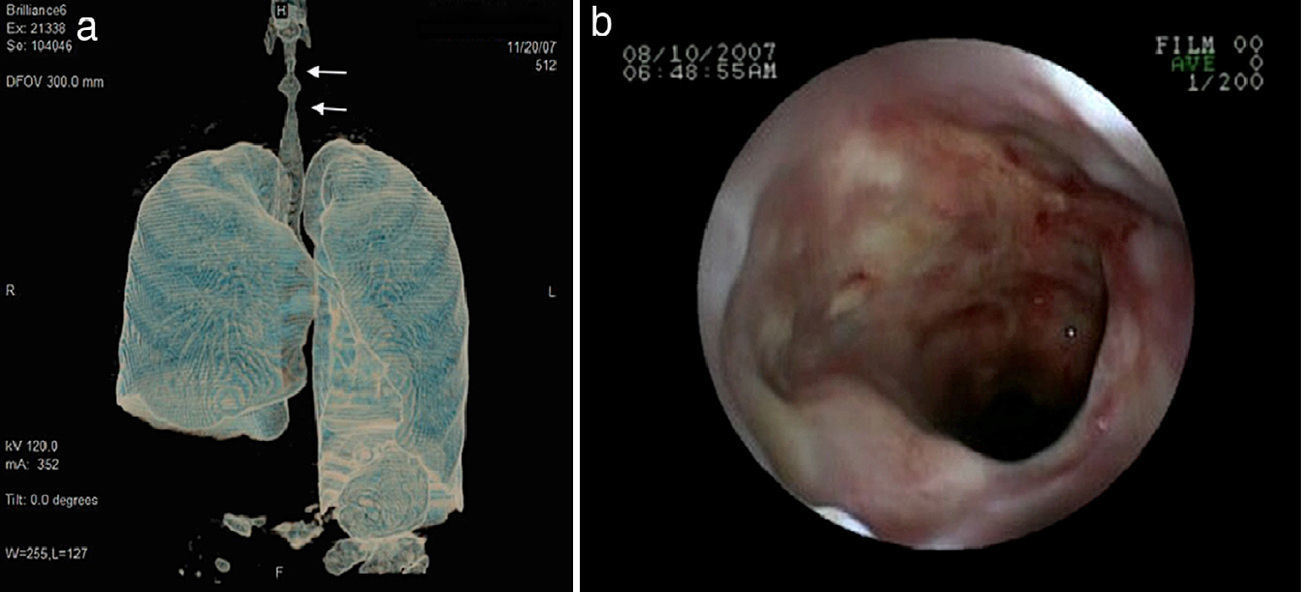
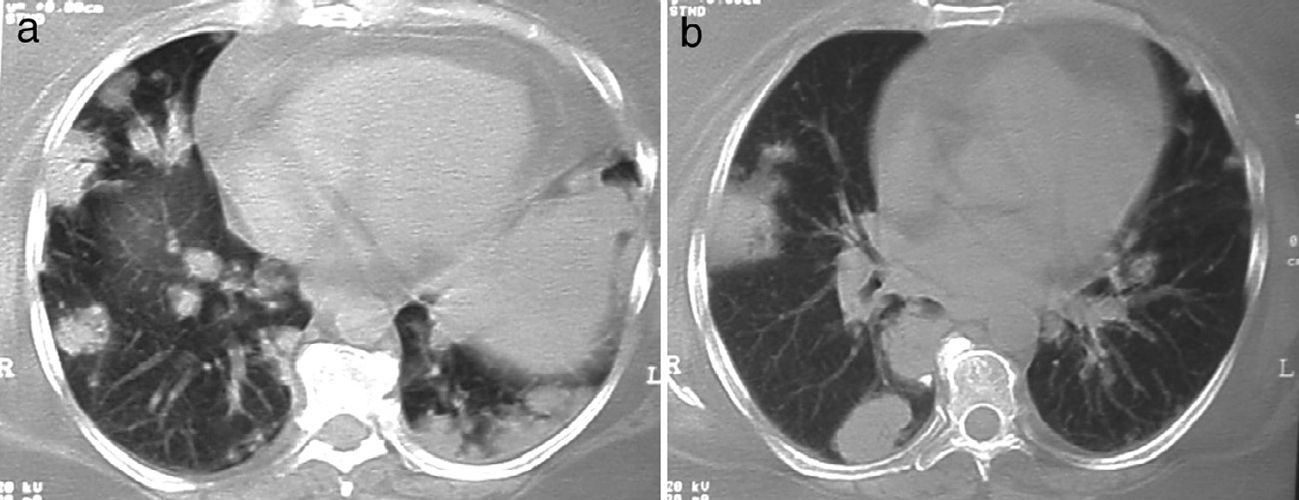
Lung nodules (LN) were the most frequent finding related to respiratory disease, present in 9 patients (64%) (Fig. 1a and b). The PN had well-defined borders and were distributed similarly in upper and lower lobes. The number of nodules per patient ranged from 2 to 20, with a mean of 12. The diameter of the nodules/masses ranged from 10 to 75mm. In one patient a halo sign was observed in some nodules. In 5 of the 9 patients, the PN were cavitated, with thick walls and irregular inner edges, without a liquid level therein; in 3 cases the PN were the only pulmonary abnormality and in 6 it was accompanied by other abnormalities, such as pulmonary consolidation in 6 (42%) (Fig. 2), frosted glass or widespread patches in 3 (21%) (Fig. 2), a mosaic pattern in 2 (14%) (Fig. 1) and centrilobular nodules in 2 (14%) (Fig. 1). Other thoracic manifestations were subglottic tracheal stenosis with involvement of respiratory mucosa without lesions (Fig. 3a and b), chronic pneumonitis and cavitation in upper and posterior segments of the right lower lobe with 2 cavitated non-adjacent pulmonary nodules (fig. 2a and b), both with negative ANCA. One patient had lung masses and PN with no cavitation in the lung biopsy organizing pneumonia and granulomatous vasculitis with necrosis (Fig. 4a and b). Only two patients had data suggestive of decreased caliber of the lobar bronchi CT with normal bronchoscopic findings and no evidence of reduction in the diameter of the lumen of these bronchi.
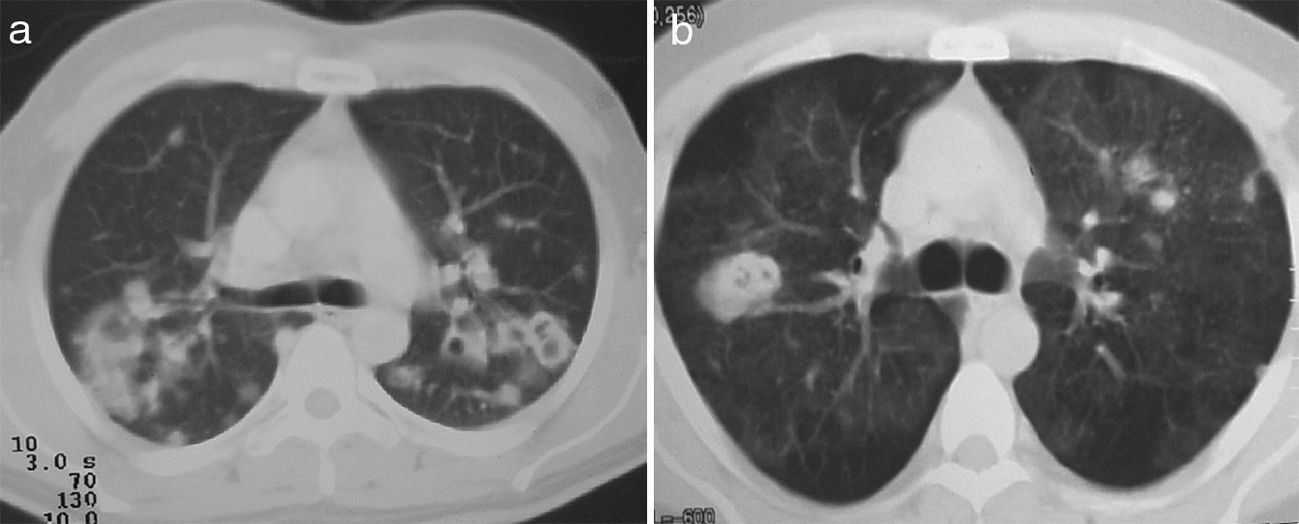
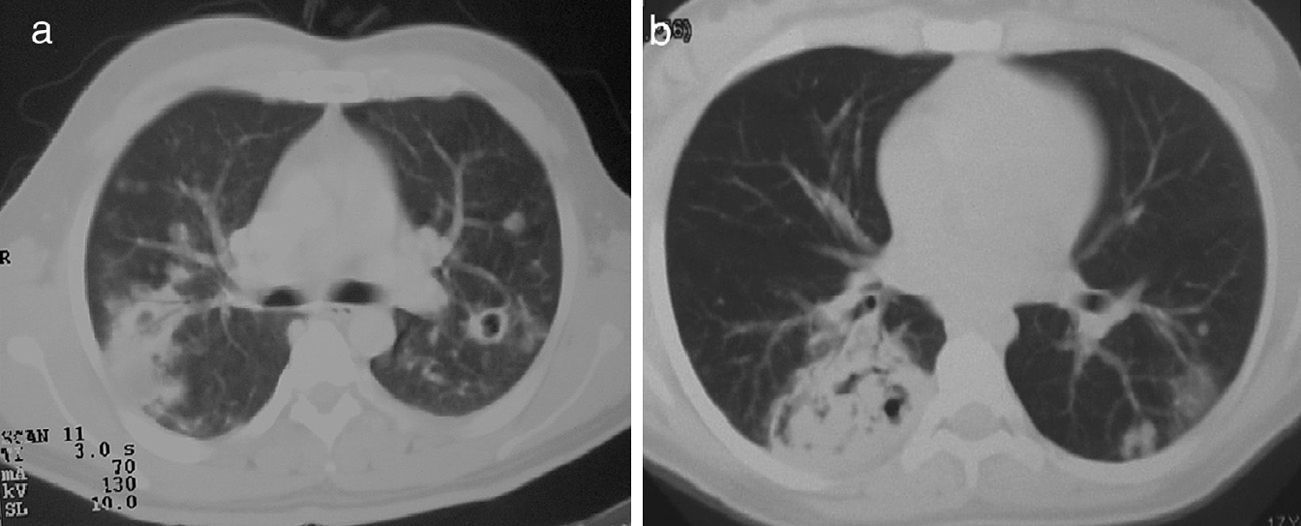
Mediastinal isolated lymphadenopathy was seen in 3 patients (23%), left or right paratracheal and subcarinal, with an average diameter of 11mm. Three patients had discreet pleural thickening in posterior regions of the lower third of the chest. Two patients had alveolar filling and frosted glass images compatible with diffuse alveolar hemorrhage and confirmed by bronchoalveolar lavage; one was associated with PN (Fig. 1a) without acute hypoxemic respiratory failure, another required invasive mechanical ventilation and presented rapidly progressive renal failure.
Seven patients received cyclophosphamide, 6 monthly cycles (1g/dose iv) and were maintained with azathioprine (100–150mg/day) due to severe kidney disease at diagnosis or during progression, and the remaining patients were treated with azathioprine and/or methotrexate (17.5mg/week), which varied during progression due to treatment response and/or adverse events; 3 patients received adjunctive therapy with azathioprine and trimethoprim-sulfamethoxazole, and all received steroids at different doses during the course of the disease.
Follow UpA control tomographic scan was performed after 6–12 months of immunomodulatory therapy in 11 patients, 8 of them with pulmonary nodules; 3 patients were followed at other hospitals, and data were not available. In 5, there was disappearance of the nodes, and in the remaining ≥3 evidence of a decrease in 90% of their volume (Fig. 5A). All patients had linear opacities or bands retracting the parenchyma or pleura in some areas where nodules (Fig. 5b) were found previously. In no case was there persistent cavitations. Areas that had frosted glass images had normal follow-up. In the case of subglottic tracheal disease, progression was to complete stenosis despite the tracheal wall infiltration with steroids and balloon dilatation.
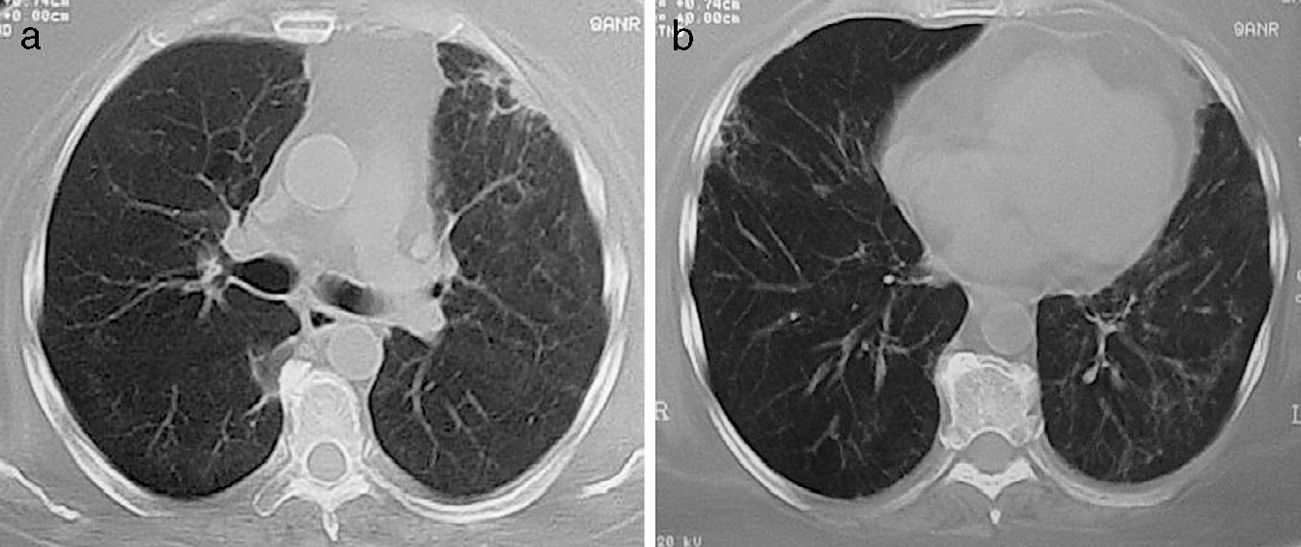
(a and b) Same patient as in Fig. 4 after 4 months of treatment with immunomodulators. Focal fibrous bands and patches of fibrosis remain, with the disappearance of the masses and nodules.
Six of the 11 patients who were followed for at least one year had a recurrence of the disease (54%) when decreasing treatment, 5 of them with pulmonary nodules and one with alveolar hemorrhage, with a similar radiological pattern in 4 of them. One patient had a “rattle” appearance on the inside of a peripheral cavitation on CT at 12 month follow-up, suggestive of fungoma, which an open biopsy showed to be necrotizing granulomatous vasculitis without evidence of fungi or mycobacteria; a year later this same patient had recurrence of multiple nodules and extensive subpleural cavitation (diameter of 6cm) in the right lower lobe; bronchoscopy with bronchoalveolar lavage and biopsy of the wall of the cavitation showed acid-fast bacilli; the patient showed improvement after adding anti-tuberculosis treatment drugs to the immunomodulatory treatment.14
DiscussionA cohort of GPA patients with respiratory disease and diverse initial clinical manifestations is presented. This series, although consisting of a small number of patients, compiles much of the thoracic manifestations described in the literature associated with this disease,3–5,8–13 and most cases have a histologic diagnosis and have post-treatment control CT.
The degree of respiratory disease and the average number of months between the onset of symptoms and diagnosis indicates the importance of the internist or specialist in respiratory diseases familiar with respiratory imaging and clinical manifestations of this disease, both for a diagnosis as for early treatment. Because of nonspecific symptoms, the radiological presentation and the diversity of its clinical expression, it is not uncommon to have other diagnoses to be more prevalent.
The frequency of PN was 64%, half of which were cavitated, corresponding with those reported in the literature (65%–70% and 50%, respectively).3,8–13 Since PN are the main imaging finding in GPA, the differential diagnosis is made primarily with infectious processes, mycobacteria and fungi, metastatic solid tumors, other granulomatous processes and bacterial endocarditis. PN were present in 6 of the 9 patients, presenting other opacities; lung consolidation typically representing pulmonary infarction and hemorrhage,15 ground-glass opacity resulting from alveolar hemorrhage, infiltration of alveolar septa with necrosis or a perfusion pattern altered by vasculitis of small vessels 15, the mosaic pattern and centrilobular nodules which may translate arteriolar disease.16
It is not recommended that the diagnosis of GPA is sustained only on ANCA positivity in isolation10 but rather these are one element in a systemic disease compatible with GPA. The specificity of ANCA for GPA depends heavily on the pre-test clinical probability, given that the disease is rare in our area, it is advisable to rule out other more prevalent and comorbid conditions, such as infectious granulomatous processes.10,17,18
Other abnormalities present in imaging studies have been reported with low frequency, similar to the findings of the present cohort: pleural thickening, mediastinal or hilar lymphadenopathy, 5% and 15%, respectively.8–11 Diffuse alveolar hemorrhage, often caused by ANCA-associated vasculitis and present in 10% of cases of GPA, shows a characteristic clinical and radiological pattern of acute hypoxemic respiratory failure with diffuse ground glass appearance or consolidation of both lungs, and is often associated with kidney disease.19 It is advisable to perform differential diagnosis with other conditions that produce this syndrome, such as systemic lupus erythematosus, microscopic polyangiitis, neumorrenal disease associated with anti-glomerular basement membrane antibodies, antiphospholipid syndrome and granulomatous polyangiitis with eosinophilia (Churg-Strauss), along with some comorbidities, such as uremic pneumonitis and heart failure. The clinical history, bronchoscopy and BAL for microbiological analysis, renal biopsy (to rule antibody deposition and verify glomerular disease), ANA, anti-GBM and ANCA are useful tools for definitive diagnosis.10
Of interest are some cases with thoracic manifestations, observed in patients with lung masses whose histology revealed organizing pneumonia and adjacent necrotizing granulomatous vasculitis, a fact that has been previously described by Oikonomou et al.,20 and represents the diverse inflammatory response of different lung pathologies with overlapping histological patterns which may reflect the host response and pose difficulties in the interpretation, but which are usually helpful to explain the alterations observed through imaging studies. Cavitary pneumonia with adjacent nodes, an unusual presentation of GPA, also represents a diagnostic and therapeutic challenge because the association of adverse events related to treatment includes infections that could resemble the disease.14
The radiological changes found in the follow up of patients have been described in other series and are also interpreted as characteristic.5,11–13 In the majority of cases, nodules disappear without a trace, and other lesions leave scarring.
It is important to point out the diagnostic difficulties of finding isolated cavitations with irregular internal walls, particularly in countries with a high prevalence of TB, a diagnosis that must be considered both at the beginning of systemic vasculitis as after administration of immunomodulatory therapy.21,22 It is recommended that invasive studies, either bronchoscopy including biopsy or open biopsy, be performed to rule out infectious complications (fungi and mycobacteria) in the context of an immunosuppressed patient.14,21
The patient with subglottic stenosis did not respond to medical and interventional management; proposed therapy in these cases is not always useful, with a limited number of cases described in the literature and no response to systemic therapy, although isolated cases have been described that respond to steroid infiltration of the mucosa and balloon dilatation,23,24 something that was ineffective in our patient. The affection of the tracheobronchial tree occurs from 15% to 30% in various series,8–10 usually observed as thickening of the bronchial wall, whether main or lobar as well as reduction of the caliber of its lumen.
This cohort is representative of GPA with variable lung expression of systemic vasculitis, which usually represents a diagnostic and therapeutic challenge to both the internist and pulmonologist, as well as for the rheumatologist. The approach requires a critical analysis of clinical, radiographical, biomarkers, cultures and histological studies through bronchoscopy or a lung biopsy by thoracoscopy, at the beginning and not rarely on follow up under immunomodulatory therapy.
The limitations of this cohort are the small number of cases, which could reflect the proper reference system for these patients. Furthermore, the reason that all patients had clinical respiratory disease is largely related with the fact that the included patients were referred to the service of respiratory diseases.
Ethical ResponsibilitiesProtection of people and animalsThe authors declare that this research has not performed experiments on humans or animals.
Data privacyThe authors declare that they have followed the protocols of the workplace on the publication of data from patients and all patients included in the study have received sufficient information and gave written informed consent to participate in the study consented.
Right to privacy and informed consentThe authors have obtained informed consent from patients and/or subjects referred to in the article. This document is in the possession of the corresponding author.
Conflict of InterestThe authors have no conflicts of interest.
Please cite this article as: Gómez-Gómez A, Martínez-Martínez MU, Cuevas-Orta E, Bernal-Blanco JM, Cervantes-Ramírez D, Martínez-Martínez R, et al. Manifestaciones pulmonares de la poliangeítis granulomatosa. Reumatol Clin. 2014;10:288–293.


