



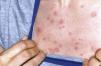

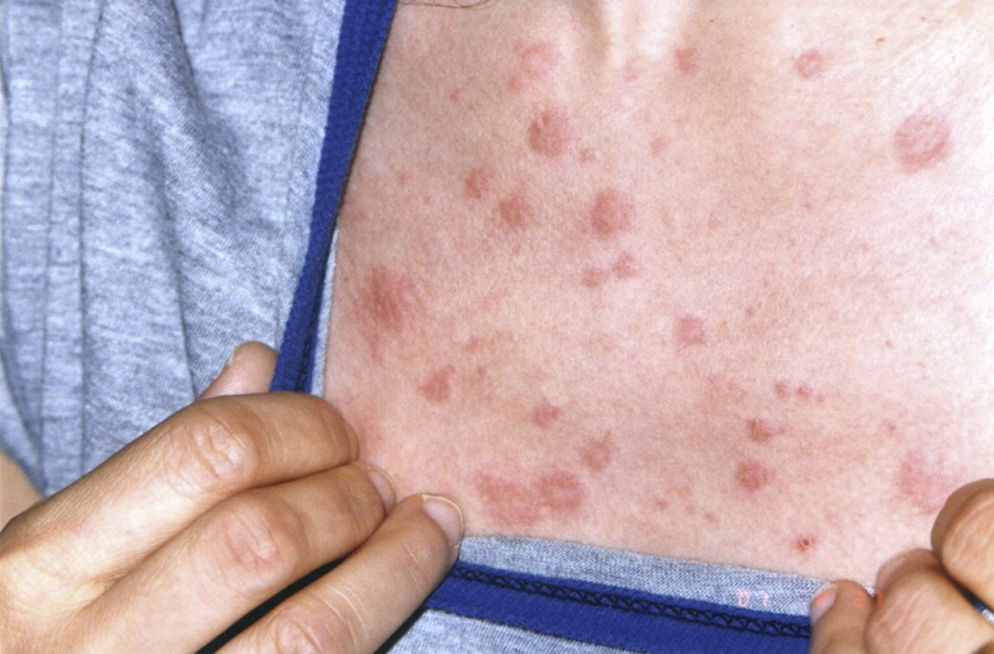
Schnitzler syndrome, described in 1974, is an autoimmune chronic urticaria syndrome associated with a characteristic monoclonal IgM component, in addition to fever, joint pain and lymphadenopathy.1
Several authors have reported patients with urticaria, fever, joint pain and increased erythrocyte sedimentation rate (ESR) and an IgG monoclonal component, suggesting that this could be a variant of Schnitzler syndrome. Clinical manifestations do not appear to differ between the typical disease and its variants. The diagnostic criteria can therefore be extended to include the variant IgG.2–4 We report the case of a 38-year-old woman first seen in 2001, who presented a persistent IgG monoclonal component and maculo-papular erythematous lesions, non-painful or itchy, at trunk level (Fig. 1) and upper limbs, as well as perforation of the nasal septum. During her progression, she developed diarrhea accompanied by abdominal pain, paresthesias, hepatosplenomegaly, livedo reticularis, bone pain and rapidly progressive sensorineural hearing loss. Laboratory tests showed a non-regenerative anemia, ESR 100mm/h and a gamma monoclonal IgG band 3580mg/dl (vn: 600–1650), Bence-Jones proteinuria (−), ANA-HEp2 (−), normal complement and normal anti neutrophil cytoplasmic antibodies (ANCA) cytoplasmic (cANCA) and perinuclear pattern (pANCA), TSH, T3, T4 and thyroid antibodies, with a negative Hansen test. Abdominal subcutaneous fat biopsy: congo red negative. X-rays of the skull, pelvis, dorso-lumbar spine, chest, mento-naso and fronto-naso were normal, a normal tomography of the abdomen with contrast, and an electromyography of the 4 extremities showing an axonal, asymmetric and distal neuropathy. Skin biopsy observed necrotizing leukocyte vasculitis. Given the different hematological findings, a new bone marrow aspirate was performed with a negative cytogenetic study for lymphoproliferative diseases. All other causes of monoclonal gammopathy (collagen disease, amyloidosis, Hansen, POEMS and neoplasms) were ruled out leading to the diagnosis of Schnitzler's syndrome (Lipsker criteria), diagnosed in 2009 (Table 1).5 This syndrome can be mimicked by other diseases such as cryoglobulinemia, urticarial hypocomplementemic vasculitis, acquired C1 inhibitor deficiency, hyper-IgD syndrome and adult Still's disease. In 2010, the patient presented anemia, increased plasma cells in the bone marrow (25%), increased IgG and positive Bence-Jones proteinuria again, with flow cytometry showing a heterogeneous group of plasma cells which expressed an intense CD 138 CD 38+ (+) CD19 (−), CD 56 (+) (±4.20% of total cells) immunophenotype, which corresponded to atypical plasma cells, leading to the diagnosis of multiple myeloma. We must consider, in the differential diagnosis, other entities characterized by monoclonal gammopathy and chronic urticaria, namely amyloidosis, chronic auto-inflammatory syndromes: Muckle–Wells or Sweet syndrome and neoplasms. Other symptoms that may be present include hearing loss, chronic inflammatory demyelinating polyneuropathy, headache, depression, dizziness, peripheral neuropathy associated with anti-AMG (anti-myelin associated glycoprotein), thrombophilia, antiphospholipid syndrome and hyperhomocysteinemia. The overall prognosis of Schnitzler syndrome depends on the possible progression to a lymphoproliferative disorder (15%–20%), either lymphomas, including lymphoplasmacytic lymphoma, Richter type lymphoma, marginal zone lymphoma, myeloma or Waldenstrom's disease. The latter may occur 10–20 years after the onset of symptoms. The patient underwent chemotherapy without an adequate response. She is currently awaiting a bone marrow transplant.
Criteria for the Diagnosis Schnitzler Syndrome.
| Major criteria (both are required): chronic urticarial dermal rash and monoclonal gammopathy (IgM or IgG) | Minor criteria (at least 2): intermittent fever, joint pain or arthritis, bone pain, palpable lymphadenopathy, splenomegaly or hepatomegaly, elevated ESR, leukocytosis and bone abnormalities (X-ray or histological) |
The treatment of this disease is difficult and disappointing. The remarkable effect of inhibition of IL-1 has led to new expectations for these patients.5–7 There is currently no Anakinra available in Argentina.
The interest of this article lies in the presence of a monoclonal IgG variant, together with the clinical and histological lesions and the subsequent progression to multiple myeloma.
Please cite this article as: Gallo J, Paira S. Síndrome de Schnitzler. Reumatol Clin. 2015;11:124–125.


