

La dermatomiositis es un tipo de miopatía inflamatoria idiopática que afecta al músculo esquelético y a la piel. Los objetivos de esta revisión son, en primer lugar, dar una pincelada breve sobre las manifestaciones cutáneas de la enfermedad para los que no estén tan familiarizados con éstas; en segundo lugar, plantear algunas cuestiones aún debatidas sobre la dermatomiositis amiopática, y, en tercer lugar, revisar un aspecto muy interesante como es el de la dermatomiositis y el riesgo de desarrollo de neoplasia. Respecto a este último punto, está claro que los pacientes con dermatomiositis tienen un mayor riesgo de desarrollo de una neoplasia y si bien no hay marcadores clínicos o biológicos claros de la presencia de ésta, es posible que en un futuro próximo se pueda disponer de autoanticuerpos que puedan desempeñar este importante papel.
Dermatomyositis is a form of idiopathic inflammatory myopathy that involves skeletal muscle and skin. The objectives of this review are to briefly describe the cutaneous manifestations of the disease, to raise some questions still debated about amyopathic dermatomyositis, and to reflect current knowledge of an interesting aspect in dermatomyositis as it is the risk to develop malignancy. Although clear evidence for a significant dermatomyositis-cancer association exists, optimal clinical or biological factors that predict an association with cancer have not been identified. Recently, some specific autoantibodies in dermatomyositis have been shown to be associated with internal malignancy. They open up the possibility to have available serological markers for detecting cancer-associated myositis in the near future.
La dermatomiositis es una enfermedad inflamatoria que afecta a la piel y el músculo. Se incluye dentro de las miopatías inflamatorias idiopáticas o miositis idiopáticas, que son un grupo heterogéneo de enfermedades musculares de etiología desconocida que se caracterizan por la aparición progresiva de debilidad muscular e inflamación1.
ClasificaciónCon la intención de ordenar estos procesos se han propuesto varias clasificaciones. Una primera clasificación clínica es la que distingue grupos específicos de miopatías inflamatorias que difieren en la clínica, la microscopia, el pronóstico y, probablemente también, la etiopatogenia (tabla 1)2. En este sentido, las más actuales incluyen, además de las entidades que propusieron Bohan y Peter en 19753, otras miopatías descritas con posterioridad, como la miopatía por cuerpos de inclusión o situaciones clínicas que si bien los dermatólogos reconocían desde hace tiempo no se contemplaban en estas clasificaciones hasta hace pocos años. Éste es el caso de la dermatomiositis amiopática o dermatomiositis sine miositis; un diagnóstico de gran interés para el clínico dada su repercusión en el pronóstico y en la terapéutica.
Tabla 1. Clasificación clínica de las miopatías inflamatorias idiopáticas
| Polimiositis |
| Dermatomiositis |
| Dermatomiositis sine miositis |
| Polimiositis y dermatomiositis de la infancia |
| Miopatía por cuerpos de inclusión |
| Miositis asociada a cáncer |
| Miositis asociada a conectivopatías |
| Miositis eosinofílica |
| Miositis granulomatosa |
| Miositis focal o nodular |
| Miositis ocular u orbital |
Una segunda clasificación contempla la presencia de unos anticuerpos dirigidos, en su mayor parte, a las enzimas que participan en la síntesis de proteínas y que parecen definir grupos con hallazgos clínicos, epidemiológicos y pronósticos homogéneos, en especial aquéllos asociados a los anticuerpos que resultan específicos de miositis (tabla 2)4,5,6. Su sensibilidad no es muy grande, por lo que su ausencia no puede excluir el diagnóstico de miopatía inflamatoria pero su presencia sí tiene un elevado valor predictivo. Dentro de los anticuerpos específicos de miositis los más importantes son los anticuerpos antisintasa (antiaminoacil-ácido ribonucleico de transferencia [ARNt] sintasa) que van dirigidos a las enzimas citoplásmicas que catalizan la unión covalente de los aminoácidos con sus ARNt. El anti-histidil-ARNt o anti-Jo-1 es el más frecuente. Este anticuerpo antisintasa delimita junto a los anticuerpos anti-Mi-2 y los anti-partícula de reconocimiento de señal diferentes cuadros clínicos con un curso evolutivo y una respuesta al tratamiento bien conocidos (tabla 2). Los anticuerpos anti-Mi-2 se asocian a dermatomiositis, tanto juvenil como del adulto, a bajo riesgo de enfermedad pulmonar intersticial y a un relativo buen pronóstico7. Este anticuerpo reconoce 2 antígenos, Mi-2α (240kD) y Mi-2β (218kD), que probablemente pertenezcan a la misma familia de proteínas (helicasas nucleares) que tienen una función reguladora de la transcripción8,9,10. Es posible que en un futuro próximo se identifiquen diferencias clínicas entre los pacientes con distinta reactividad frente a las 2 moléculas Mi-2 o frente a determinados fragmentos de éstas, como ya se ha apuntado en un artículo reciente11. La partícula de reconocimiento de señal (PRS) es un complejo citoplásmico ácido ribonucleico-proteína que consiste en 7 SL-RNA y 6 polipéptidos de 72, 68, 54, 19, 14 y 9kD. Este complejo media la translocación de polipéptidos a través del retículo endoplásmico.
Tabla 2. Anticuerpos en las miositis idiopáticas y características clinicoevolutivas de la miopatía inflamatoria asociada *
| Anticuerpo | Antígeno | Pacientes con anticuerpos (%) | Características de los antígenos | Síndrome clínico asociado | Evolución y pronóstico |
| Anticuerpos específicos de miositis | |||||
| Antisintasas | Enzimas citoplásmicas que catalizan la unión covalente de los aminoácidos con su ARNt | Comienzo en primavera de forma aguda con miositis, artritis, afección pulmonar intersticial, fiebre, “manos de mecánico” y fenómeno de Raynaud | Moderada respuesta a tratamiento y recurrencias en su evolución. Supervivencia a los 5 años del 65% (por insuficiencia respiratoria y cor pulmonale) | ||
| Anti-Jo-1 | Histidil-ARNt sintasa | 20 | |||
| Anti-PL-7 | Treonil-ARNt sintasa | 5–10 | |||
| Anti-PL-12 | Alanil-ARNt sintasa | <5 | |||
| Anti-OJ | Isoleucil-ARNt sintasa | <5 | |||
| Anti-EJ | Glicil-ARNt sintasa | 5–10 | |||
| Anti-KS | Asparaginil-ARNt sintasa | <5 | |||
| Anti-Zo | Fenilalanil-ARNt sintasa | <1 | |||
| Anti-YRS | Tirosil-ARNt sintasa | <1 | |||
| Anti-partícula de reconocimiento de señal | Partícula de reconocimiento de señal | 5 | Complejo citoplásmico que media la translocación de polipéptidos a través del retículo endoplásmico | Comienzo muy agudo y grave en otoño, con afección muscular grave, afección miocárdica y disfagia. Miopatía necrosante | Mala respuesta a tratamiento. Supervivencia a los 5 años del 25 al 30% (por afección cardíaca) |
| Anti-Mi-2 | Helicasa nuclear (218/240kD) | 5–10 | Helicasa nuclear con función reguladora de la transcripción | Comienzo agudo y leve con las lesiones cutáneas clásicas | Buena respuesta a tratamiento. Supervivencia a los 5 años del 95% |
| Anti-CADM-140 | Desconocido (proteína 140kD) | 50 con DMA | Desconocida | Específico de la DMA | |
| Anti-p155/p140 | TIF-1-γ | 20 con DM | DM, sobre todo asociada a cáncer | ||
| Anti-MJ | Desconocido (proteína 140kD) | <5 | DM juvenil | ||
| Anti-PMS1 | Enzima reparadora de AND | <5 | |||
| Anticuerpos asociados a miositis | |||||
| Anti-U1 ribonucleoproteína | U1 ribonucleoproteína nuclear | 10 | Miositis overlap, enfermedad mixta de tejido conectivo | ||
| Anti-Ku | Subunidad reguladora ADN-PK (70/80kD) | 20–30 | Síndrome overlap polimiositis-esclerodermia en japoneses | ||
| Anti-PM-Scl | Complejo nuclear de 11 a 16 proteínas | 8–10 | Síndrome overlap polimiositis-esclerodermia en caucásicos |
ADN: ácido desoxirribonucleico; ADN-PK: ácido desoxirribonucleico dependiente de proteincinasa; ARNt: ácido ribonucleico de transferencia; DM: dermatomiositis; DMA: dermatomiositis amiopática; TIF-1-γ: factor transcripcional intermediario 1-γ.
* Adaptada de Mimori T et al 5 .
Los pacientes en los que se desarrollan anticuerpos frente a esta partícula (anti-PRS) suelen presentar una miositis de inicio agudo, en general resistente al tratamiento estándar con glucocorticoides y con frecuentes exacerbaciones, afectación miocárdica y disfagia12,13. Desde el punto de vista microscópico, se ha observado que en esta miositis predomina la necrosis de las fibras musculares casi sin existencia de infiltrados inflamatorios14. Por tanto, los anticuerpos anti-PRS pueden ser el marcador de un síndrome de miopatía necrosante diferente de la típica polimiositis que, si bien no suele responder al tratamiento convencional con glucocorticoides, sí puede responder a otros tratamientos como rituximab (anticuerpo monoclonal anti-CD20)15. Como se verá más adelante, otros autoanticuerpos de más reciente identificación parecen asociarse a otras situaciones clínicas dentro del contexto de la dermatomiositis.
Manifestaciones cutáneasAdemás del músculo, la piel tiene un papel protagonista en esta enfermedad. Las lesiones cutáneas resultan muy características y preceden o son concomitantes al desarrollo de la miositis en un porcentaje elevado de pacientes, lo que en muchas ocasiones permite al dermatólogo ver por primera vez al paciente y realizar el diagnóstico de la enfermedad.
De esta erupción destaca la tonalidad violácea de las lesiones y su distribución alrededor de los ojos y en las prominencias óseas que forman el exantema heliotropo y las pápulas del signo de Gottron, respectivamente. A esto debe añadirse para completar el cuadro típico la intensa afectación de la cutícula ungueal con engrosamiento de ésta y la presencia de pequeños infartos hemorrágicos16,17,18,19.
El exantema heliotropo, llamado así por su característica coloración violácea, casi siempre afecta los párpados de forma bilateral y simétrica y suele acompañarse de un cierto grado de edema. No debe confundirse la inusual situación clínica en la que el edema resulta tan intenso que lleva a la formación de verdaderas ampollas. Tampoco debe pasar desapercibido un eritema tan tenue que únicamente se presente como un leve cambio de coloración de la piel en el borde de los párpados.
Ante un edema y un eritema palpebral que es asimétrico y no tiene una tonalidad violácea se deben plantear otros diagnósticos, como un lupus eritematoso profundo, una oftalmopatía tiroidea o un seudotumor orbitario, entre otros20 (tabla 3).
Tabla 3. Diagnóstico diferencial del edema o del eritema palpebral unilateral
| Agudo | Crónico |
| Infecciones oculares | Linfedema |
| Picadura de insecto | Tumor palpebral u orbitario (primario o secundario) |
| Eccema de contacto | |
| Urticaria | Dermatosis granulomatosas |
| Infecciones cutáneas | Lupus eritematoso profundo |
| Trombosis del seno cavernoso | Seudotumor orbitario |
| Parálisis facial | |
| Algias vasculares de la cara | Esclerodermia en banda |
| Blefarochalasia | |
| Oftalmopatía tiroidea |
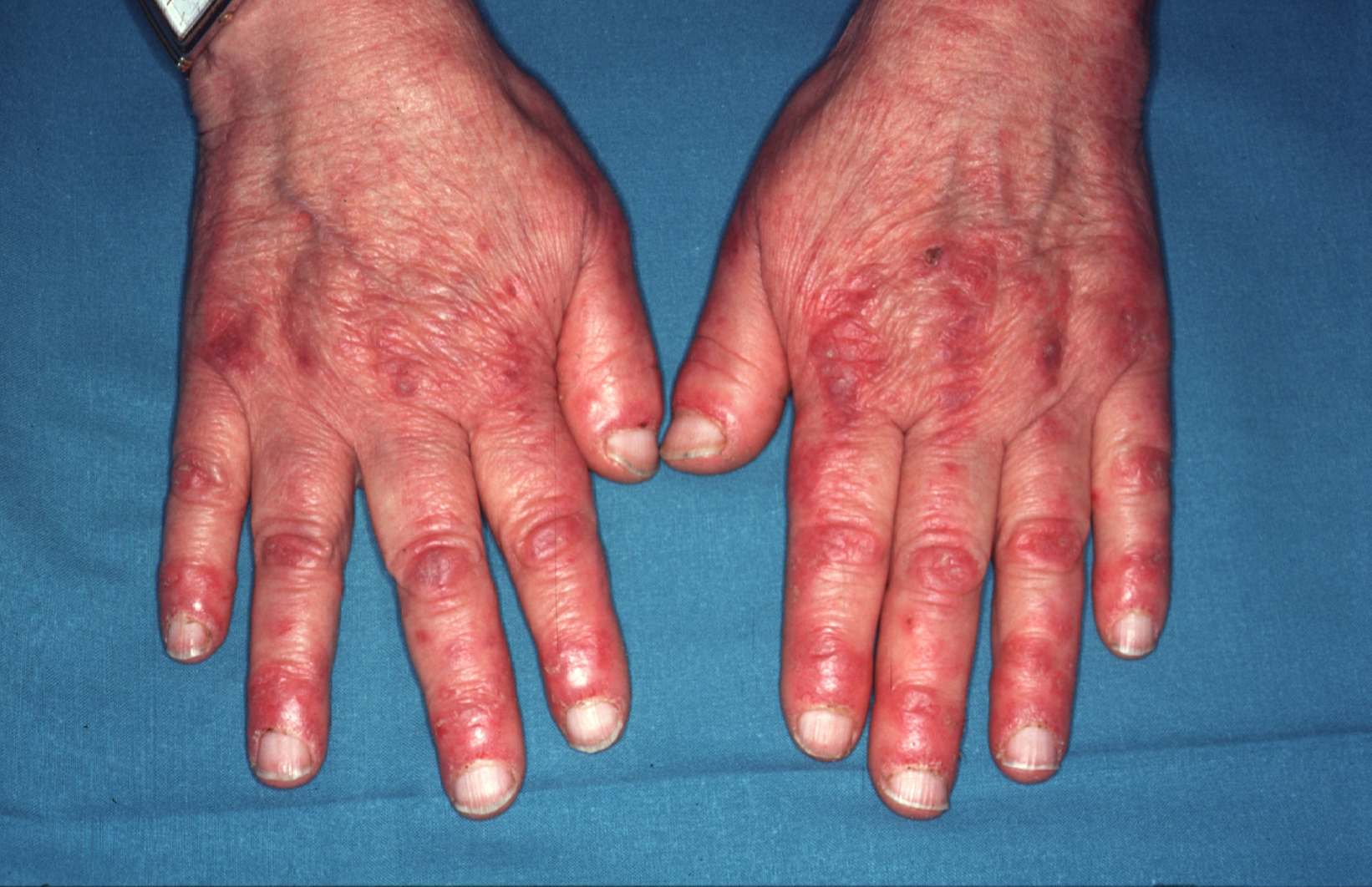
El signo de Gottron hace referencia a unas pápulas y placas algo violáceas acompañadas de descamación leve o, en ocasiones, de descamación prominente de tipo psoriasiforme, que se asientan sobre todo en las prominencias óseas, en especial sobre las articulaciones metacarpofalángicas y las articulaciones interfalángicas (figura 1). También pueden aparecer sobre los codos, las rodillas o cualquier otra articulación. Estas lesiones pueden confundirse en la clínica con un lupus eritematoso, una psoriasis o un liquen plano. En estas 2 últimas enfermedades el análisis microscópico de la biopsia es de gran ayuda19.
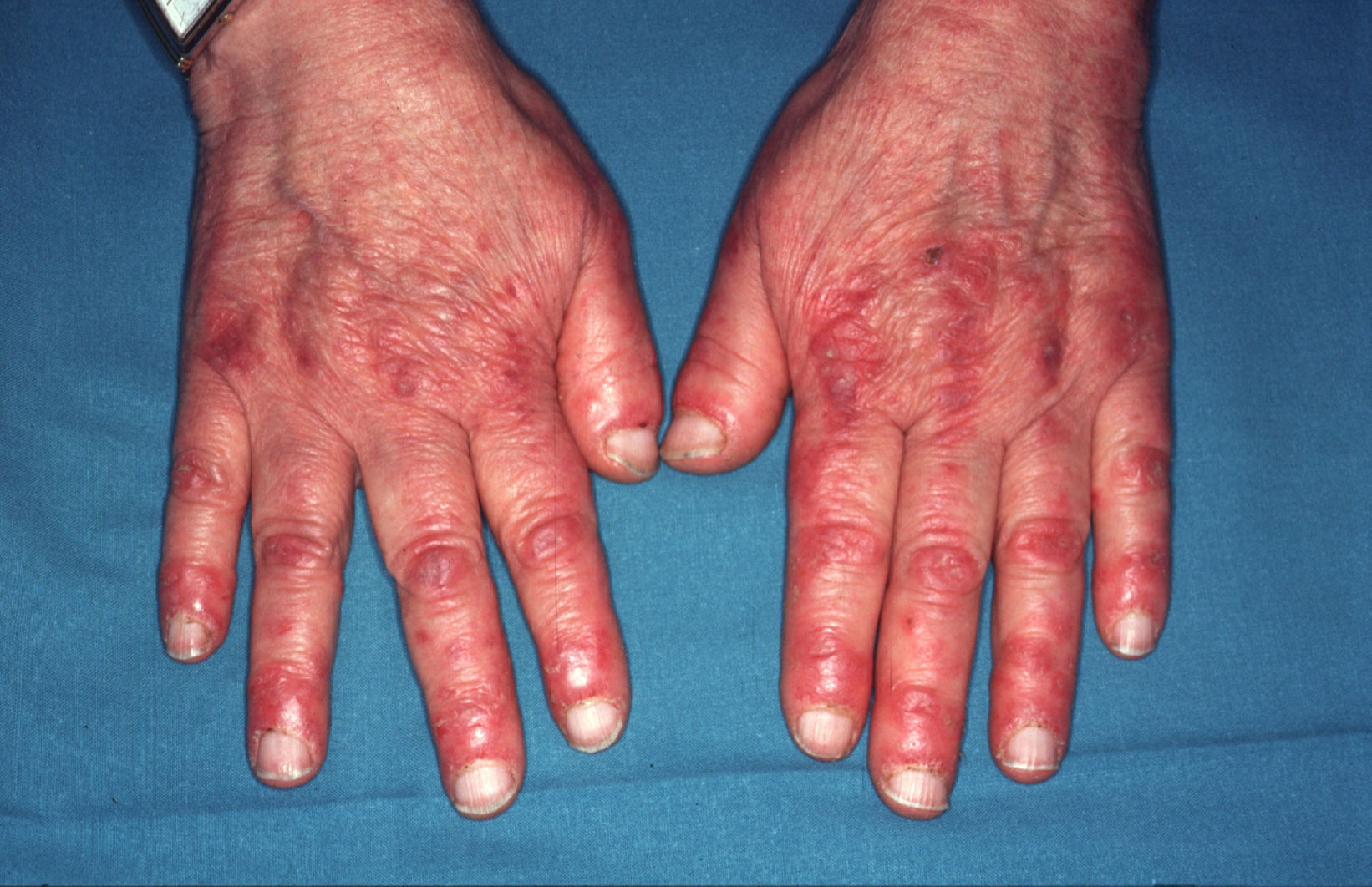
Figura 1. Eritema en el dorso de las manos que predomina sobre los nudillos y en la zona periungueal característico de la dermatomiositis.
En el caso del lupus eritematoso, con cambios microscópicos que pueden ser muy similares a los de la dermatomiositis, un pequeño detalle como la localización de las lesiones puede ser de gran interés. En el lupus eritematoso, cuando las lesiones asientan en el dorso de las manos, éstas se hallan en los espacios interdigitales y respetan los nudillos.
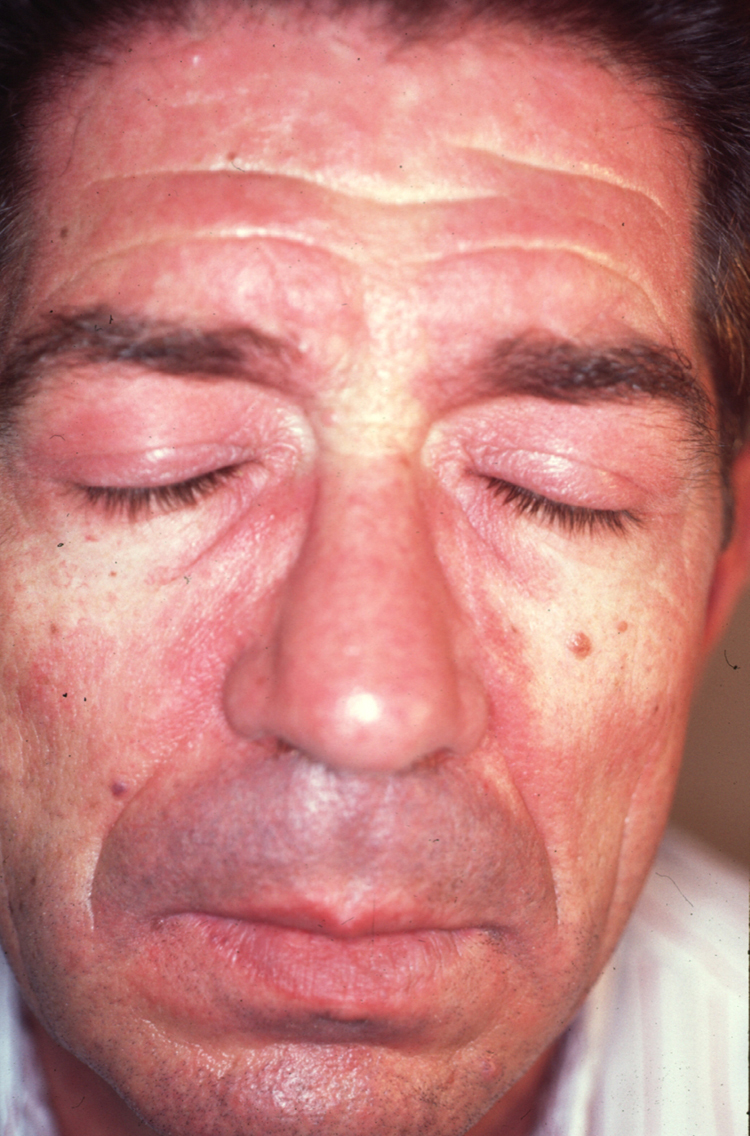
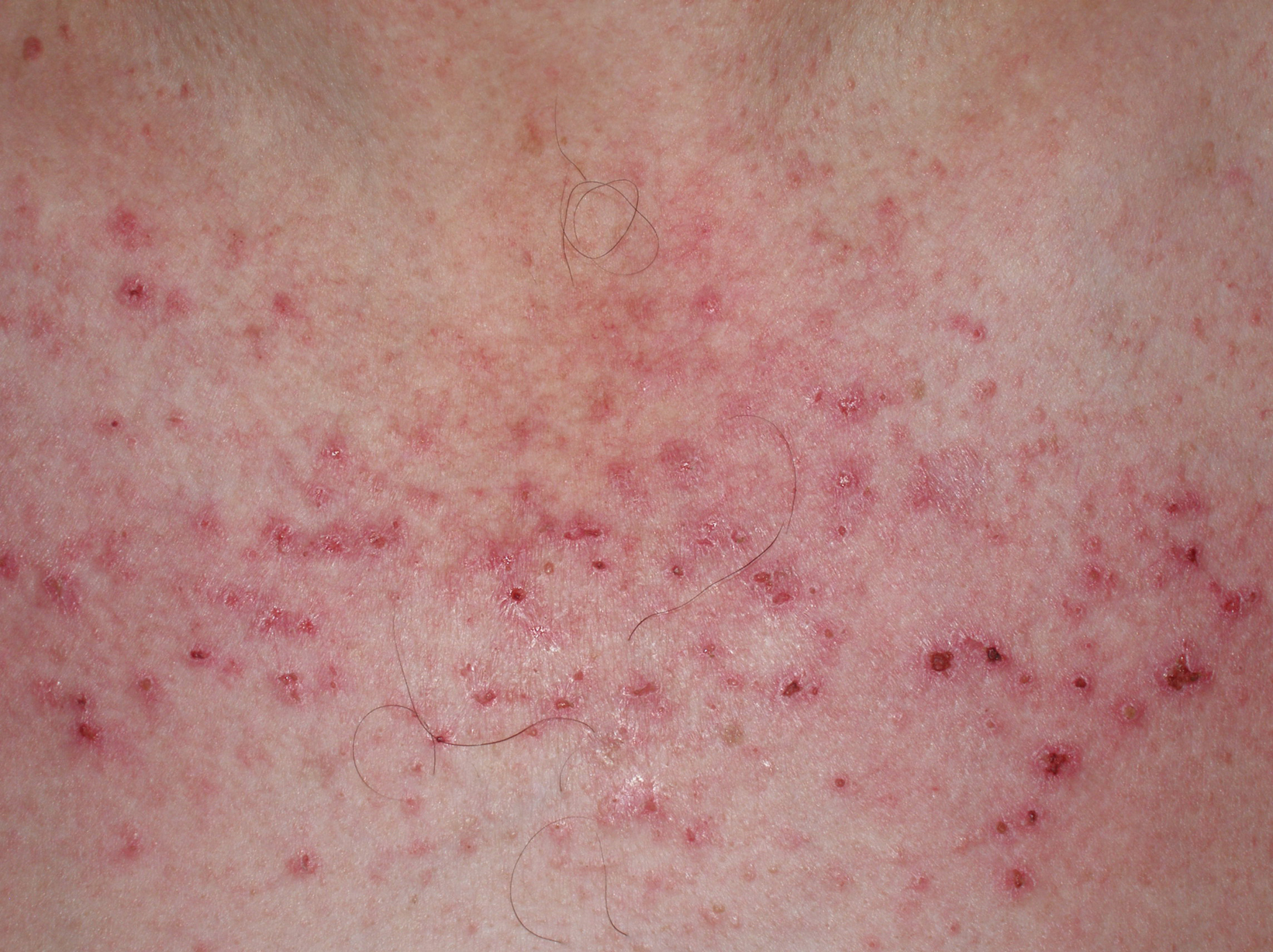
A partir de estas localizaciones, patognomónicas o muy características, el eritema puede extenderse al resto de la cara (figura 2) y ocupar fundamentalmente la zona central o áreas seborreicas, el cuero cabelludo, el tronco (sobre todo en la cara anterior del cuello y la «V» del escote), la nuca, los hombros y el tercio superior de la espalda, lo que configura el clásico eritema en capelina o la superficie de extensión de las extremidades. Sobre el eritema, no es infrecuente la aparición de cambios de poiquilodermia (se entiende como ésta la presencia de pequeñas áreas de atrofia con telangiectasias y trastornos de la pigmentación)19. En ocasiones pueden verse ulceraciones o costras que indican la existencia de necrosis de la piel por isquemia (figura 3) (v. apartado Dermatomiositis y neoplasia).
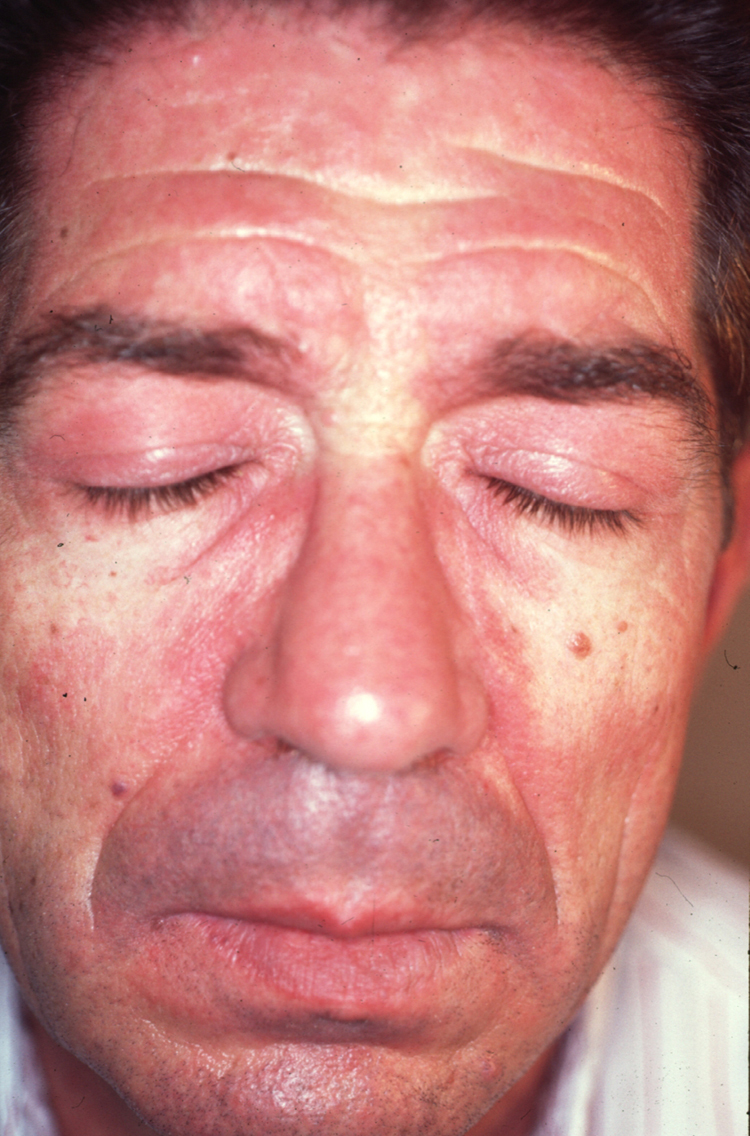
Figura 2. Eritema facial de tonalidad violácea que predomina en las áreas seborreicas. Nótese también la afectación de los parpados que configuran al exantema heliotropo.
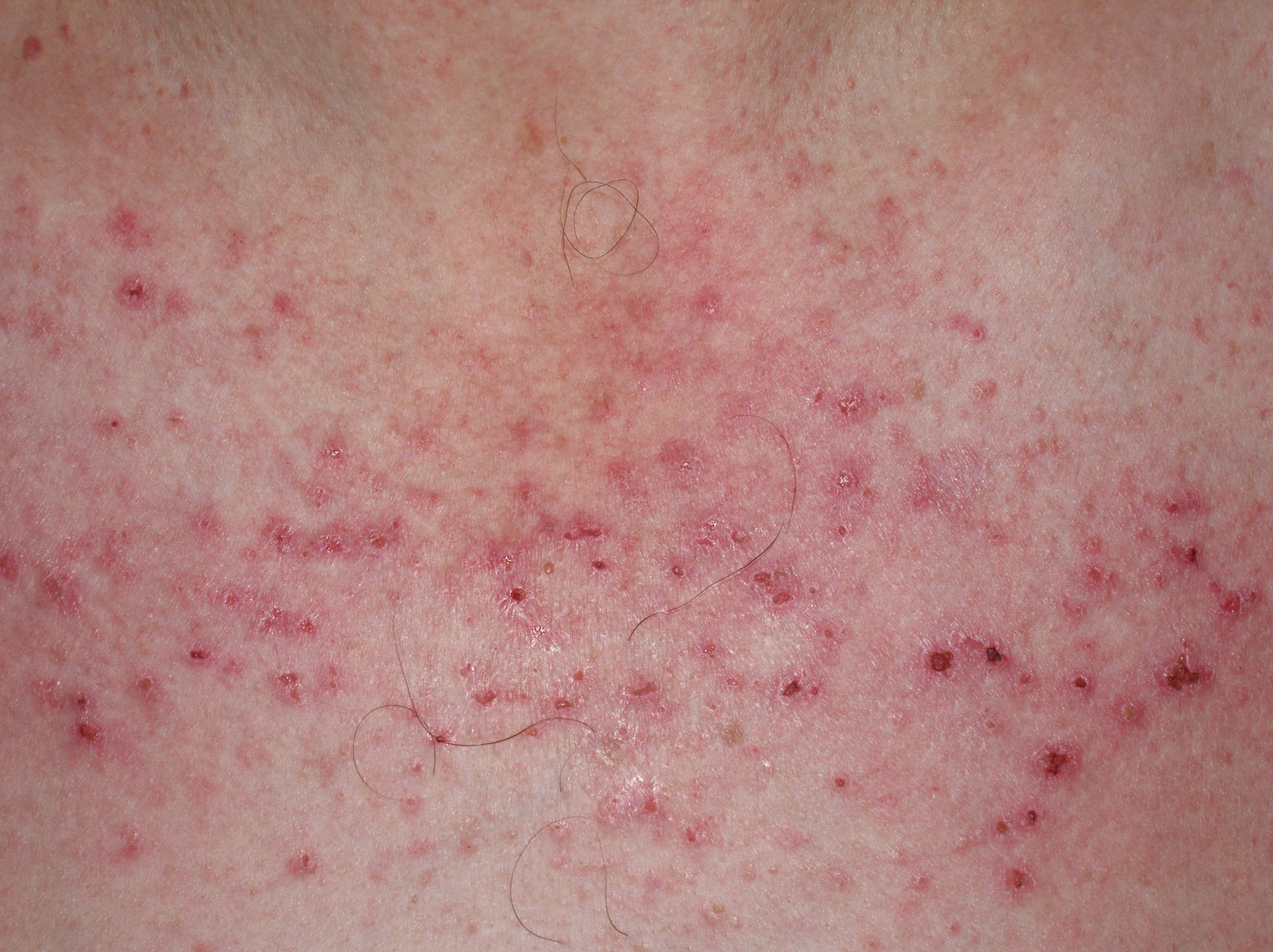
Figura 3. Lesiones eritematocostrosas en el escote de un paciente afectado de una dermatomiositis. Las lesiones eritematocostrosas son secundarias a la existencia de isquemia capilar y, según algunos autores, su presencia es indicativa de asociación a neoplasia.
La calcinosis de la piel y el músculo es infrecuente en el adulto pero puede ocurrir hasta en el 40% de los niños y adolescentes con dermatomiositis. La calcinosis cutánea se manifiesta en forma de nódulos duros, amarillentos o del color de la piel que se asientan con frecuencia sobre las prominencias óseas. En ocasiones, los nódulos pueden abrirse al exterior con riesgo, entonces, de infección secundaria. La calcificación en los músculos suele ser asintomática y puede ser sólo un hallazgo radiológico. En los casos graves, la calcinosis puede ser causa de limitación funcional de los miembros afectados19,21.
Otras lesiones que se han descrito como muy infrecuentes en la dermatomiositis son las lesiones vesiculoampollares22, la paniculitis23 y una curiosa erupción denominada eritema flagelado24,25. Esta última consiste en la aparición de unas bandas lineales no pruriginosas, persistentes, que se asientan en el tronco y las extremidades de un paciente que, por otro lado, presenta además las lesiones cutáneas típicas de dermatomiositis. Los pacientes niegan el rascamiento de la piel y no es posible demostrar en ellos la existencia de un dermografismo. Las bandas lineales parecen seguir una distribución centrípeta y sin solución de contigüidad con las áreas de eritema del tercio superior de la espalda o del escote. Estas lesiones, similares a las que pueden presentar los pacientes que reciben bleomicina, no se han observado en ninguna otra conectivopatía autoinmune, por lo que se consideran muy características de la dermatomiositis. Si bien no hay una explicación plausible para el desarrollo de esta peculiar erupción, se acepta que es una manifestación clínica más de esta enfermedad desde el momento en que en la microscopia se han demostrado cambios similares a los ya descritos en las lesiones típicas de la dermatomiositis.
La presencia de mucina en la dermis es un hallazgo microscópico frecuente en las lesiones cutáneas de dermatomiositis. Sin embargo, sólo en ocasiones se ha descrito la traducción clínica de estos depósitos especialmente intensos en forma de pápulas o de placas violáceas infiltradas al tacto, localizadas en el tronco, las extremidades o que sigan los pliegues de flexión de las palmas y los dedos26,27. Estas lesiones pueden ser la única manifestación cutánea de la enfermedad, por tanto, siendo entonces el diagnóstico especialmente difícil.
Finalmente, en los pacientes con dermatomiositis se pueden ver otros cambios, como engrosamiento, hiperqueratosis y fisuras en las caras lateral y palmar de los dedos de las manos, que configuran las llamadas “manos de mecánico”. Estos cambios muy curiosos y llamativos se describieron inicialmente en pacientes afectados de diversas conectivopatías (enfermedad mixta del tejido conectivo, dermatomiositis y lupus eritematoso sistémico) pero con el denominador común de presentar una miositis28. Más tarde, se observaron asociados a la presencia de enfermedad pulmonar intersticial, miositis, artritis y enfermedad de Raynaud (en un porcentaje variable), todo esto bajo el denominador común de la presencia en sangre periférica del anticuerpo anti-Jo-1. Esto justificó el término “síndrome antisintasa” acuñado para identificar a los pacientes que cursaban con esta sintomatología29,30. Finalmente, las “manos de mecánico” se han descrito junto a lesiones típicas de la dermatomiositis clásica, en pacientes con polimiositis30 o en síndromes overlap, como la escleromiositis31. Por todo esto, en la actualidad las “manos de mecánico” se consideran un marcador cutáneo de miositis con independencia del anticuerpo o de la miopatía asociada.
Dermatomiositis sin miositisEn un porcentaje bajo de pacientes, que se ha descrito entre el 2 y el 18%, se puede desarrollar una erupción indistinguible de la dermatomiositis clásica pero con ausencia o mínima expresión de enfermedad muscular. A este grupo se le denomina dermatomiositis sine miositis, dermatomiositis amiopática o dermatomiositis clínicamente amiopática como recientemente han propuesto Sontheimer et al32.
Esta situación clínica es motivo de gran interés y controversia ya que siguen sin definirse los criterios que deben regir su diagnóstico. Asimismo, se desconoce si supone el mismo riesgo que la dermatomiositis clásica de desarrollar complicaciones graves, como una neoplasia o una enfermedad pulmonar intersticial, ni hay acuerdo sobre qué tratamiento es el más adecuado.
Respecto a su diagnóstico, se debe recordar que no hay diferencias en cuanto a la clínica y a la microscopia de las lesiones cutáneas en la dermatomiositis amiopática respecto a la dermatomiositis clásica y que en más del 50% de los pacientes con una dermatomiositis clásica las lesiones cutáneas preceden entre 3 y 6 meses a la afectación muscular33. Se acepta que si ésta ocurre durante los primeros 2 años desde la aparición del exantema debe considerarse como la progresión habitual de la dermatomiositis clásica. Pasado este tiempo, si el paciente sigue con enfermedad únicamente en la piel, puede hablarse de dermatomiositis sine miositis34,35.
Resulta más controvertido definir la ausencia de enfermedad muscular y hasta qué punto debe explorarse el músculo para decidir si este órgano está activo o no. Se ha observado que pacientes con lesiones cutáneas de dermatomiositis sin clínica de debilidad muscular y valores de CK (creatin kinase ‘creatincinasa’) dentro de la normalidad pueden tener alteraciones en el electromiograma, la resonancia magnética y la biopsia muscular, lo que implica la existencia de una miositis clínicamente asintomática36,37. Sin embargo, como se demuestra en un estudio reciente en el que se realiza una revisión sistemática de la literatura médica32, estos hallazgos no sirven para predecir el inicio posterior de enfermedad muscular franca y, por tanto, no necesariamente comportan una actitud terapéutica más intensiva. Así, se puede argumentar que más allá de la exploración de la clínica muscular y la determinación de la CK, otras exploraciones musculares en ausencia de debilidad muscular resultarían innecesarias con el fin de tomar una decisión terapéutica. El inicio de la enfermedad muscular se anuncia con frecuencia por una elevación de la CK, lo que subraya el interés de realizar determinaciones periódicas de esta enzima muscular en los pacientes con una dermatomiositis clínicamente amiopática, en especial, durante los primeros 2 años.
Es evidente que ante la dificultad de definir desde el punto de vista clínico la dermatomiositis amiopática, la caracterización de algún marcador serológico que permitiera identificar a estos pacientes tendría un gran interés clínico y pronóstico. En este sentido, en los pacientes con dermatomiositis clínicamente amiopática y no en la dermatomiositis clásica se han identificado algunos anticuerpos dirigidos a nuevos autoantígenos que podrían desempeñar este papel; entre estos destaca el anticuerpo anti-CADM-140 que se dirige a un antígeno citoplásmico de 140kD y se asocia, al menos en la población japonesa, a la dermatomiositis amiopática y, dentro de este contexto, a enfermedad pulmonar rápidamente progresiva38. Respecto a esta última complicación, la revisión de la literatura médica permite afirmar que hasta en el 15% de las dermatomiositis clínicamente amiopáticas se puede desarrollar enfermedad pulmonar intersticial con una mortalidad de casi el 40% de los casos32. Hasta la identificación de anti-CADM-140 también se había podido comprobar que en estos pacientes, a diferencia de lo que ocurre en la dermatomiositis clásica asociada a enfermedad pulmonar, no se identificaba ningún anticuerpo específico de miositis clásico, como el anticuerpo anti-Jo-1.
En lo referente a la actitud terapéutica, Euwer y Sontheimer demostraron en 199139 que una aproximación más intensiva en el tratamiento de la enfermedad cutánea (se entiende como la actitud que se plantearía en el caso de una dermatomiositis clásica) podría prevenir el desarrollo posterior de inflamación muscular. Sin embargo, la publicación posterior de varias series en las que en los pacientes diagnosticados de dermatomiositis amiopática no se desarrolló enfermedad muscular a pesar de no recibir tratamiento inmunosupresor37,40,41 sugiere que los glucocorticoides orales u otros agentes inmunosupresores sólo deberían administrarse en presencia de enfermedad muscular abierta.
Dermatomiositis y neoplasiaEn 1916 se demostró por primera vez la asociación entre dermatomiositis y neoplasia. En los primeros estudios epidemiológicos, diversos aspectos clínicos y metodológicos impidieron la confirmación42,43,44,45,46,47,48,49. Entre estos aspectos se encuentran la dificultad en el diagnóstico de miositis y, sobre todo, la distinción entre dermatomiositis y polimiositis, el sesgo en la referencia de los casos, los estudios con muestras pequeñas, la escasa duración del seguimiento o la ausencia de un grupo control apropiado, entre otros. Sin embargo, estudios de cohortes más recientes y bien diseñados han demostrado una asociación significativa entre miositis y neoplasia; el riesgo de esta asociación es mayor para la dermatomiositis que para la polimiositis.
En el año 2001 un grupo australiano publicó uno de estos estudios en el que se incluyó a un total de 537 pacientes, todos ellos con biopsia probada de miopatía inflamatoria50. La razón de incidencia estandarizada que se obtuvo en el grupo de pacientes con dermatomiositis fue de 6,2, lo que indica que el riesgo de neoplasia es 6 veces mayor en este proceso que en la población general. También se observó que este riesgo fue 2,4 veces mayor en los pacientes con dermatomiositis que en los pacientes con polimiositis.
Además, Hill et al51 demostraron en un amplio grupo de pacientes con dermatomiositis y polimiositis que ambos se asociaban a un mayor riesgo de cáncer, pero este riesgo era mayor en los pacientes con dermatomiositis que en los pacientes con polimiositis.
Por tanto, parece claro que hay una asociación entre dermatomiositis y cáncer. Sin embargo, sigue sin definirse cuál es la estrategia más adecuada en la búsqueda de esta neoplasia. En este sentido, 3 cuestiones importantes no tienen aún una respuesta clara. Primero, ¿se dispone de algún factor predictivo o de algún marcador de neoplasia en la dermatomiositis del adulto?; segundo, el estudio de la neoplasia ¿debe realizarse con las mínimas exploraciones o, por el contrario, debe ser completo y exhaustivo?, y tercero, ¿durante cuánto tiempo se debe realizar el seguimiento del paciente en caso de que no se encuentre neoplasia alguna en la primera evaluación?
La primera cuestión es una de las más importantes para el clínico, ya que es obvio que la identificación de determinados hallazgos clínicos o parámetros biológicos que pudieran actuar como marcadores de neoplasia permitiría realizar una investigación selectiva y meticulosa de la neoplasia sólo en aquellos pacientes que fueran portadores de estos marcadores. Sin embargo, lamentablemente hay pocos cambios clínicos que permiten sospechar la existencia de una neoplasia. El primer factor que se puede considerar es la edad. Se sabe que la frecuencia de neoplasia en los pacientes con dermatomiositis se incrementa con la edad42,52,53,54,55,56 y que la presencia de neoplasia resulta excepcional en la dermatomiositis infantil19. No obstante, se ha demostrado que el riesgo de neoplasia se encuentra aumentado incluso en los pacientes con dermatomiositis por debajo de los 45 años. Por tanto, la edad no debería ser para el clínico un elemento de disuasión en la búsqueda meticulosa de la neoplasia.
Algunos autores han señalado en diversas publicaciones de la literatura médica francesa57,58,59 el interés de observar en los pacientes con dermatomiositis lesiones necróticas en la piel, ya que parece que pueden ser un marcador de neoplasia. En uno de estos estudios el valor predictivo de la asociación de la necrosis cutánea y de la neoplasia fue del 70%59. Este parámetro clínico resulta fácil de evaluar para el dermatólogo y probablemente su observación podría justificar una investigación exhaustiva de la neoplasia.
Finalmente, se ha mostrado que la presencia de enfermedad pulmonar intersticial sola o acompañada de anticuerpos antisintasa tiene una asociación negativa con el riesgo de cáncer4,60,61.
Respecto a los parámetros biológicos, se puede distinguir entre algunas determinaciones de laboratorio habituales, los marcadores de antígenos tumorales y los autoanticuerpos. Se ha descrito que los pacientes con dermatomiositis y neoplasia asociada tienen con mayor frecuencia unos valores de CK normales62,63,64 aunque para algunos autores sería lo contrario65,66 y una velocidad de sedimentación globular incrementada65.
Es bien conocido que la determinación de una serie de marcadores de antígeno tumoral puede proporcionar una información útil antes de iniciar la búsqueda exhaustiva de la neoplasia. En los pacientes con miositis puede resultar de especial interés los marcadores CA 125 y CA 19.9, ya que según Amoura et al61 la elevación en el suero de estos 2 marcadores así como la elevación del marcador CA 125 en determinaciones seriadas se asocia a un mayor riesgo de desarrollo de cáncer, no sólo de ovario sino también de otros tipos de cáncer.
Desde el punto de vista serológico, hasta hace muy pocos años no se había identificado ningún anticuerpo específico de miositis como marcador de neoplasia. Más aún, en la literatura médica se había señalado que la presencia de estos anticuerpos específicos de miositis disminuía la probabilidad de cáncer4,67. No obstante, en los últimos años se han identificado nuevos anticuerpos específicos en los pacientes con dermatomiositis38,68 y alguno de éstos parece que se asocia a la presencia de cáncer. Uno de estos anticuerpos es el anticuerpo contra la proteína de 155kD (anti-p155). Según Targoff et al69, el anticuerpo anti-p155 estuvo presente en el 75% de los casos con miositis y neoplasia y la neoplasia se desarrolló en el 37,5% de los pacientes con dermatomiositis y el anticuerpo anti-p155 positivo. El antígeno diana de este anticuerpo es el factor transcripcional intermediario 1-γ70. Otros autores, de forma casi simultánea, han descrito un anticuerpo similar que reacciona no sólo con una proteína de 155kD sino también con otra proteína de 140kD71,72. Esta doble banda de precipitación se mencionaba en el estudio anterior69, por lo que probablemente se trate del mismo anticuerpo. En cualquier caso, estos 2 grupos de trabajo71,72 identifican este anticuerpo como específico de dermatomiositis a la vez que se correlaciona bien con la presencia de neoplasia y la ausencia de enfermedad pulmonar. Así, un anticuerpo anti-p155/140 positivo proporciona una alta especificidad (96%), una sensibilidad moderada (50%) y un valor predictivo negativo elevado (97%) para dermatomiositis asociada a cáncer. Más aún, la presencia de este anticuerpo junto a un panel negativo de anticuerpos específicos de miositis incrementa la sensibilidad (94%) y el valor predictivo negativo (99%)72. Es obvio que la aplicación clínica de todos estos resultados requiere su confirmación mediante estudios amplios con un seguimiento prospectivo, pero evidentemente abre la posibilidad de disponer en un futuro próximo de posibles marcadores serológicos de dermatomiositis asociada a neoplasia.
Respecto a la segunda cuestión planteada, sigue siendo motivo de discusión cómo debe realizarse el estudio de una neoplasia en el paciente con una dermatomiositis. Se debe partir de la certeza de que el tipo de cáncer que se puede encontrar es variado (los más frecuentes son los de ovario, pulmón, tracto gastrointestinal, páncreas y mama) y que la mayoría de éstos pueden estar ocultos51. Desde una perspectiva clínica, parece razonable aconsejar al paciente asintomático la búsqueda de cualquier proceso cuya detección y tratamiento precoces conduzcan a mejorar la evolución de éste. Por otro lado, es evidente que la presencia de una neoplasia es un indicador de mal pronóstico en el contexto de una dermatomiositis.
Tradicionalmente, hay 2 actitudes opuestas respecto al número y tipo de exploraciones que se deben realizar en la búsqueda de una neoplasia en esta enfermedad. Una de éstas se limita a realizar un interrogatorio completo, una exploración física minuciosa, determinaciones de laboratorio habituales y exploraciones complementarias en función de los síntomas y de los signos que se desprendan de la historia clínica. La otra aproximación incluiría, además de los exámenes ya comentados, un amplio abanico de exploraciones, como tomografía computadorizada (TC) toracoabdominal, endoscopia del tracto gastrointestinal, mamografía, biopsia de médula ósea e inmunoelectroforesis del suero, entre otras65,73. Pero probablemente todas estas recomendaciones no puedan verse como algo definitivo sino que pueden variar en el tiempo de acuerdo con la introducción de nuevos conocimientos médicos y de técnicas de exploración más sensibles y cómodas. Por ejemplo, aquí puede plantearse el papel de la tomografía por emisión de positrones.
Hoy por hoy, y según un estudio reciente de Hill et al51, parece razonable que a un paciente varón de origen caucásico con una dermatomiositis se le realice además de un examen clínico cuidadoso, los análisis habituales y los marcadores de antígeno tumoral, una búsqueda de sangre en heces y una TC toracoabdominal. En la mujer, también se justificaría la realización de una TC y una ecografía pélvica, así como una mamografía. Los estudios endoscópicos del tracto intestinal superior e inferior pueden indicarse de acuerdo con la edad del paciente54,55,65. Finalmente, en los pacientes asiáticos que residen en el sudeste de Asia el cáncer de nasofaringe es muy frecuente, por lo que en ellos sería recomendable además una cuidadosa evaluación del territorio otorrinolaringológico64,73.
Aunque se han descrito algunos casos aislados de cáncer asociado a dermatomiositis amiopática, no hay datos poblacionales que confirmen el aumento del riesgo de neoplasia en este subtipo de dermatomiositis32. No obstante, se recomienda la misma actitud vigilante respecto a la posibilidad de una neoplasia asociada.
Entre el 26 y el 70% de los casos el desarrollo de la neoplasia ocurre durante el primer año tras el diagnóstico de la miositis. Este dato confirma que la investigación meticulosa de una neoplasia debe realizarse sobre todo durante este período de tiempo. Sin embargo, diversos estudios han demostrado que el riesgo es más alto en los 3 primeros años, pero continúa alto a los 5 años tras el diagnóstico de miositis. No se puede descartar que este riesgo tardío de neoplasia se deba a un efecto a largo plazo del tratamiento inmunosupresor. En cualquier caso, es recomendable que el clínico al cuidado de pacientes con dermatomiositis realice una supervisión meticulosa anual en búsqueda de una posible neoplasia durante los 3 o 4 primeros años tras el inicio de la miositis50,51.


