









Las miopatías inflamatorias idiopáticas son un grupo heterogéneo de enfermedades cuya principal característica es la debilidad muscular y la identificación de una inflamación subyacente en la biopsia muscular. Se incluyen en este grupo la dermatomiositis, la polimiositis y recientemente la miositis con cuerpos de inclusión, con toda probabilidad la menos inflamatoria y también la miopatía adquirida más frecuentemente a partir de los 50 años. Aunque el principal órgano diana es el músculo, la piel y el pulmón, entre otros órganos internos, se afectan con frecuencia, por lo que las miopatías inflamatorias se consideran enfermedades sistémicas. En ocasiones pueden asociarse a cáncer y la presencia de autoanticuerpos específicos y asociados a estas enfermedades sustenta la etiología autoinmune del proceso y ayuda a categorizar a los pacientes. El tratamiento incluye la administración de glucocorticoides, inmunodepresores y puntualmente terapias biológicas, sin descuidar la rehabilitación incluso en la fase aguda de la enfermedad.
Idiopathic inflammatory myopathies are a group of heterogeneous, acquired systemic diseases characterized by progressive symmetrical muscle weakness, elevated serum levels of muscle enzymes, electromyographic abnormalities, and inflammatory infiltrates on muscle biopsy. Characteristic histopathologic features allow classification of idiopathic inflammatory myopathies into polymyositis, dermatomyositis, and sporadic inclusion-body myositis. These are commonly regarded as autoimmune disorders, and various autoantibodies directed to specific nuclear and cytoplasmic antigens are found. Other organs besides the muscle can be involved being the skin and lung the most frequent. Occasionally dermatomyositis and polymyositis can be associated with cancer in a paraneoplastic manner. Corticosteroids and immunosuppressive agents are the mainstay therapy, although in refractory cases biologic therapy can be used. Physical therapy can not be forgotten.
Las miopatías inflamatorias constituyen un grupo de enfermedades que se caracterizan por afectar preferentemente a la musculatura estriada y por su naturaleza inflamatoria1–3. La biopsia muscular identifica un infiltrado inflamatorio que, según su localización y distribución, contribuye decisivamente al diagnóstico. Bajo este concepto se agrupan fundamentalmente tres entidades4,5, la dermatomiositis, enfermedad bien definida; la polimiositis, que agrupa diversos trastornos que cursan con inflamación muscular y que se considera un diagnóstico de exclusión, y finalmente la miositis con cuerpos de inclusión (MCI)6,7, catalogada como esporádica, ya que hay una forma familiar indistinguible de ésta. La MCI se incorpora al grupo en los años noventa y si bien tiene un indudable protagonismo clínico, para algunos autores sólo podría incluirse en el grupo de forma tangencial, ya que el infiltrado inflamatorio que se detecta en la biopsia muscular parece más un epifenómeno acompañante de los cuerpos de inclusión característicos de esta entidad que no la verdadera causa de la debilidad muscular en estos pacientes.
Las miopatías inflamatorias, especialmente la dermatomiositis y la polimiositis, se consideran enfermedades sistémicas, ya que aunque el principal órgano diana es el músculo estriado, otras estructuras, como la piel o el sistema articular, se afectan con frecuencia. También los órganos internos, en especial el pulmón, forman parte del espectro clínico de estas enfermedades. Ocasionalmente, y sobre todo la dermatomiositis, puede asociarse a cáncer, y presenta un comportamiento paraneoplásico.
En cuanto a su frecuencia, pueden considerarse dentro del grupo de enfermedades raras debido a su baja incidencia. Estudios epidemiológicos llevados a cabo en diversos puntos del globo establecen una incidencia anual media de 2,1 a 7,7 casos nuevos por millón de habitantes y año8. En España la incidencia anual media es similar, de 2,2 a 10,6 casos nuevos por millón de habitantes y año9.
Criterios diagnósticos y diagnóstico diferencialLos criterios diagnósticos establecidos por Bohan et al1 siguen siendo de utilidad en la práctica clínica diaria por su simplicidad, así como la clasificación propuesta por estos autores, que divide a las miopatías inflamatorias en cinco grupos que incluyen la dermatomiositis y la polimiositis del adulto, la dermatomiositis infantil, la paraneoplásica y la de superposición. Esta clasificación tiene el inconveniente de no incluir la MCI, desconocida entonces, pero por el contrario permite establecer grupos de diferente pronóstico10.
La biopsia muscular es para algunos autores la prueba de referencia en el diagnóstico de las miopatías inflamatorias4,5,11. Esto, sin duda, es cierto en el caso de la dermatomiositis y en menor medida en la MCI, pero no en la polimiositis, que sigue siendo un diagnóstico de exclusión. La atrofia perifascicular por fenómenos de microisquemia y necrosis por disminución de los capilares musculares, junto a un infiltrado inflamatorio perivascular y perimisial con predominio de linfocitos T CD4+ y células B, es típica de la dermatomiositis. Técnicas de inmunohistoquímica permiten identificar el complejo de ataque de membrana del complemento (C5b9) como la causa de esta destrucción capilar. Tan característica es la biopsia en los pacientes con dermatomiositis, que puede llegarse al diagnóstico únicamente por el estudio histológico aun en ausencia de exantema cutáneo, propio de esta enfermedad. Sin embargo, la biopsia muscular puede ser negativa ya que la afectación es parcheada y el uso previo de glucocorticoides durante al menos 2 semanas puede minimizar los hallazgos patológicos. Algunos autores proponen que la práctica sistemática de una resonancia magnética (RM) permitiría realizar la biopsia en los músculos previamente identificados como alterados12.
Los hallazgos histopatológicos en la polimiositis, como la infiltración linfocítica endomisial predominantemente por linfocitos T CD8+ y el fenómeno de invasión parcial, pueden observarse en otras miopatías, principalmente en las distrofias musculares y la MCI11,13. La expresión por parte de las fibras musculares no afectadas del complejo mayor de histocompatibilidad tipo I (MHC-I) apoya el diagnóstico de polimiositis idiopática, aunque ocasionalmente puede observarse en las MCI y en alguna distrofia muscular, especialmente en las disferlinopatías. Todo ello ha llevado a cuestionar la individualización propia de la polimiositis, la convierte en un diagnóstico por exclusión y obliga a un estudio inmunohistoquímico meticuloso que descarte otras entidades como las distrofias o la MCI, especialmente en los casos resistentes al tratamiento11,12,14. Las vacuolas ribeteadas subsarcolémicas ricas en amiloide y puestas de manifiesto mediante la tinción de rojo Congo o de filamentos de amiloide en la microscopia electrónica permiten, junto al cuadro clínico característico, llegar al diagnóstico de MCI11,13,15. En las tablas 1–4 se recogen los diferentes criterios propuestos para el diagnóstico de las distintas enfermedades que forman parte del grupo de miopatías inflamatorias.
Criterios diagnósticos de la polimiositis/dermatomiositis
| Debilidad simétrica de los músculos de las cinturas escapular y pelviana, flexores del cuello, progresiva en semanas o meses, con o sin disfagia y afección respiratoria |
| Biopsia muscular característica de miopatía inflamatoria |
| Elevación enzimática muscular (creatincinasa, aldolasa, transaminasas…) |
| Hallazgos electrofisiológicos musculares característicos |
| Lesiones cutáneas patognomónicas de dermatomiositis (signo de Gottron, eritema violáceo o en heliotropo) |
| Enfermedad definida: 4 criterios; probable: 3 criterios; posible: 2 criterios. En el caso de la dermatomiositis debe cumplirse el último criterio siempre |
Tomada de Bohan et al1.
Criterios diagnósticos de dermatomiositis
|
|
|
|
HLA: antígenos leucocitarios humanos; VIH: virus de la inmunodeficiencia humana.
Criterios diagnósticos de polimiositis
|
|
|
|
AR: artritis reumatoide; EMTC: enfermedad mixta del tejido conectivo; ESP: espondiloartropatías; HTLV: virus linfotrópico de linfocitos T del ser humano; LES: lupus eritematoso sistémico; MHC: complejo mayor de histocompatibilidad; SS: síndrome de Sjogren; VIH: virus de la inmunodeficiencia humana.
Criterios diagnósticos de la miositis por cuerpos de inclusión
|
|
|
|
HTLV: virus linfotrópico de linfocitos T del ser humano; MHC: complejo mayor de histocompatibilidad; VIH: virus de la inmunodeficiencia humana.
El diagnóstico diferencial es amplio13 e incluye las enfermedades musculares hereditarias, en especial la distrofias que pueden cursar con infiltrado inflamatorio en la biopsia muscular, algunas enfermedades metabólicas, como las glucogenosis, entre las que la enfermedad de McArdle del adulto, o déficit de miofosforilasa, es la más frecuente y hay que descartarla mediante prueba de isquemia en el antebrazo16 o estudio genético; los trastornos del tiroides, que no sólo se pueden confundir con la polimiositis, sino que también pueden aparecer y coexistir con las miopatías inflamatorias condicionando una mala evolución y una respuesta terapéutica inadecuada17; las infecciones parasitarias, como la toxoplasmosis o la triquinosis, y el consumo de fármacos, en especial los fármacos utilizados en el tratamiento de las dislipemias, como las estatinas o los fibratos, pero también los glucocorticoides o los antipalúdicos de síntesis utilizados en el tratamiento de algunas manifestaciones de la enfermedad. Otro aspecto a tener en cuenta es el diagnóstico erróneo de hepatitis por elevación de las transaminasas, ya que estas enzimas también pueden ser de origen muscular y se elevan junto a la creatincinasa en las miopatías. Entre las distrofias, el déficit de disferlina, proteína de membrana de la fibra muscular y que participa en la reparación celular, es lo que con mayor frecuencia puede confundirse con una polimiositis. La sospecha y la confirmación diagnóstica de esta entidad son importantes, ya que parece que el tratamiento con glucocorticoides tiene un efecto negativo en el músculo, que empeora el cuadro clínico y la evolución18,19.
La miopatía por glucocorticoides no es rara, pero difícilmente crea un problema diagnóstico al clínico, suele ser de menor intensidad y el contexto clínico y, en caso de dudas, el estudio electrofisiológico ayudan a su identificación. En nuestra experiencia, la miopatía por glucocorticoides tiene más protagonismo clínico en las enfermedades en que el componente inflamatorio es menor o menos relevante (distrofias, MCI); predomina la toxicidad del fármaco en el músculo respecto al supuesto beneficio de actuar en la inflamación. En ocasiones, para el control de las manifestaciones cutáneas o articulares de la dermatomiositis se utilizan antimaláricos, que también son causa de miopatía, aunque con mayor relevancia histopatológica en el estudio ultraestructural que repercusión clínica20.
Manifestaciones clínicasLa forma de presentación más común de estas enfermedades es la debilidad muscular, que suele afectar de forma característica a la musculatura esquelética proximal, es decir a la cintura escapular y pelviana, dificultando las actividades que precisan del normal funcionamiento de estos músculos, como tender la ropa, peinarse, subir escaleras o levantarse de la silla, entre otras21. Esta debilidad se acompaña de una marcha miopática o anserina con oscilación de la cadera en cada paso. La musculatura facial suele estar respetada. Los músculos flexores del cuello y la musculatura estriada de la orofaringe se afectan con frecuencia; estos últimos causan la disfagia que presentan los pacientes con miositis, que en ocasiones puede ser tan intensa que se manifieste por regurgitación nasal del contenido alimentario durante la deglución y, ocasionalmente, favorecer la neumonía por broncoaspiración2,3,5. Parece que con mayor frecuencia estos pacientes presentan síndrome de apnea del sueño debido al colapso de la vía aérea superior por debilidad de la musculatura orofaríngea (observación personal, resultados no publicados). Las mialgias son un síntoma que puede aparecer, pero son poco frecuentes.
No parece haber diferencias en lo relativo a la afectación muscular entre dermatomiositis y polimiositis, pero sí en cambio en los pacientes con MCI. Suele considerarse la sospecha de esta última entidad cuando el cuadro clínico es tórpido, no responde al tratamiento convencional y, de forma característica, el paciente presenta debilidad muscular asimétrica y con afectación proximal y distal junto a gran atrofia de los músculos cuádriceps, que se traduce en caídas frecuentes, y de los flexores profundos de los dedos, que contribuye a la dificultad que tienen estos pacientes para desenroscar un tornillo o deshacer un nudo. A diferencia de la dermatomiositis y la polimiositis que predominan en el sexo femenino, como en la mayoría de las enfermedades sistémicas autoinmunitarias, la MCI es más frecuente en varones y se considera la miopatía adquirida más frecuente a partir de los 50 años5,22.
Las manifestaciones cutáneas son características de la dermatomiositis, y podemos distinguir un amplio abanico de lesiones, la mayoría de ellas con un cierto componente de fotosensibilidad, por lo que suelen aparecer en zonas expuestas al sol2,3,5. Se consideran patognomónicas el edema palpebral de color lila o en heliotropo (por ser éste el color de esta flor) y los nódulos de Gottron, áreas eritematosas discretamente descamativas e infiltradas que aparecen sobre los nudillos de las manos (figs. 1 y 2). Lesiones similares pueden observarse en zonas de extensión, como codos y rodillas, y también en la línea de inserción del cuero cabelludo y en la nuca. Otras lesiones cutáneas localizadas en la zona del escote en forma de “V” o en la espalda en forma de “chal” tienen también relación con la estimulación lumínica. En general, no se recomienda la biopsia cutánea por su inespecificidad diagnóstica. En ocasiones, las lesiones cutáneas características pueden aparecer en ausencia de afectación muscular, y cuando esta situación persiste durante al menos 2 años sin que finalmente aparezca signo alguno de miopatía, se habla de dermatomiositis amiopática, que no siempre tiene un curso benigno, ya que puede asociarse a cáncer o desarrollar una neumopatía intersticial aguda de mal pronóstico23.


Un subgrupo de pacientes presenta una característica lesión eccematosa en la zona lateral de los dedos de las manos que se denomina “manos de mecánico” (fig. 3) y que se relaciona con la presencia de anticuerpos anti-Jo-1. La paniculitis y la calcinosis (fig. 4) pueden adquirir protagonismo clínico en los pacientes con dermatomiositis, especialmente en su forma juvenil, pero también en el adulto24,25. No es infrecuente apreciar edema con fóvea en las fases agudas de la enfermedad, atribuido exclusivamente al proceso inflamatorio subyacente. La dermatomiositis y la polimiositis se consideran enfermedades sistémicas y como tales presentan las siguientes manifestaciones.
Aparato respiratorio

Es la manifestación visceral más frecuente, aparece en hasta casi la mitad de los pacientes en una u otra forma26–31. La afección respiratoria más conocida en pacientes con dermatomiositis y polimiositis es la intersticial. En general, su instauración suele ser subaguda o crónica y los hallazgos clínicos a la exploración pueden detectar estertores crepitantes secos, en “velcro”, característicos de la fibrosis pulmonar. En estos casos los anticuerpos antisintetasa, en especial los anti-Jo-1 (antihistidil-ARNt sintetasa) que son los más frecuentes, suelen ser positivos, y se constituyen en marcador de un síndrome clínico caracterizado por la presencia de miopatía inflamatoria (dermatomiositis o polimiositis), neumopatía intersticial y artritis, fiebre, fenómeno de Raynaud y “manos de mecánico”, entre otras manifestaciones menores. En ocasiones, la neumopatía intersticial adquiere el máximo protagonismo y es el motivo de consulta, la miopatía es prácticamente subclínica y el marcador inmunológico es la presencia de anticuerpos antisintetasa distintos del anti-Jo-132. El sustrato patológico que subyace en la mayoría de los casos parece ser una neumonía intersticial no específica, si bien se han descrito casos asociados a neumonía intersticial usual o neumonitis organizativa criptogénica, anteriormente conocida como bronquiolitis organizativa con neumonía organizativa focal (BONO)26,33,34. Estas formas de afectación respiratoria no parecen comportar un peor pronóstico35. La tomografía computarizada (TC) de alta resolución, el estudio inicial del lavado bronquioloalveolar y, sobre todo, de la función respiratoria que incluya la capacidad vital forzada (CVF), la difusión del CO (DLCO) y los valores de presión diafragmática inspiratorios (PIM) y espiratorios (PEM) ayudan a la valoración de estos pacientes.
Con mucha menor frecuencia, pueden desarrollar una neumonitis intersticial aguda de curso fulminante, con destrucción del parénquima pulmonar, neumomediastino y anticuerpos antisintetasa negativos, cuyo sustrato patológico es la lesión alveolar difusa y que suele conducir a una insuficiencia respiratoria irreversible en un plazo de meses a pesar de un tratamiento inmunosupresor intenso; el trasplante pulmonar es la única opción terapéutica36–39. Esta forma es más frecuente en sujetos orientales y en esta etnia se ha descrito un autoanticuerpo frente a proteínas de 140 kD que podría actuar como marcador diagnóstico de esta forma fulminante40. La musculatura respiratoria, sobre todo el diafragma, puede verse afectada también por la enfermedad28,36. En la mayoría de los sujetos hay una insuficiencia ventilatoria leve o moderada que corre paralela al curso de la enfermedad y que suele mejorar con el tratamiento de base de la miositis. La disminución de los valores de PIM y PEM es característica de esta situación y puede repercutir en el valor de la CVF, y da una falsa impresión al interpretar los valores bajos de CVF como secundarios a neumonitis intersticial. En unos pocos casos la evolución es hacia la insuficiencia respiratoria ventilatoria como consecuencia de la claudicación de la musculatura respiratoria. Mientras se espera el efecto del tratamiento inmunosupresor, la ventilación mecánica externa (BiPAP) puede actuar de puente evitando la muerte del paciente o la necesidad de intubación orotraqueal41. La hipertensión pulmonar que acompaña a la fibrosis pulmonar o de forma primaria es infrecuente, pero hay que pensar en ella en los casos que cursen con aumento de la disnea y disminución de la DLCO, a pesar de no modificarse la CVF. El estudio mediante ecocardiografía será el primer paso para llegar al diagnóstico.
Afección cardíacaEs poco frecuente, pero cuando se presenta lo hace en forma de miocarditis, comporta un mal pronóstico y puede evolucionar a miocardiopatía dilatada. En algunas series clínicas es la principal causa de muerte en estos pacientes42,43. El tratamiento convencional con glucocorticoides e inmunosupresores puede revertir el cuadro, pero en ocasiones sólo el trasplante cardíaco puede solucionar la situación44. La ecocardiografía, la RM cardíaca con gadolinio y la gammagrafía cardíaca con 99mTc son las técnicas diagnósticas más utilizadas junto al electrocardiograma42,45. Es posible que haya manifestaciones cardíacas subclínicas, así se han descrito valores elevados de la fracción miocárdica de la creatincinasa (CK-MB) en pacientes asintomáticos con miositis46. Los anticuerpos anti-SRP (single recognition particle) se han relacionado con un cuadro clínico de polimiositis en mujeres de raza negra, que cursa con afección cardíaca grave y de predominio estacional en los meses de otoño47, aunque esta asociación ha sido cuestionada.
Afectación digestivaLa disfagia que aparece en el curso de la enfermedad es debida a la miopatía de la musculatura estriada orofaríngea. Las formas de dermatomiositis juvenil pueden cursar con vasculitis intestinal y/o perforación de viscera hueca, lo que es una rareza en adultos48. La seudoobstrucción intestinal y la neumatosis cistoidea intestinal son manifestaciones poco habituales pero descritas en las dermatomiositis49,50. Se especula sobre la relación que pueda existir entre celiaquía o anticuerpos antigliadina y miopatía, especialmente en el grupo de las polimiositis y MCI51,52.
Etiopatogenia y autoanticuerposLa etiopatogenia de las miopatías inflamatorias no es bien conocida53. Sobre la base de un agente externo físico, químico o infeccioso que actúa en un territorio genético predispuesto se han avanzado algunas teorías. Un amplio estudio en diversos países del globo, sobre casi 1.000 pacientes con polimiositis o dermatomiositis, demostró que la proximidad a la latitud 0° era un factor de riesgo de dermatomiositis. Es decir, los países más cercanos a la línea del ecuador y, por tanto, más expuestos a radiación lumínica presentaban con mayor frecuencia dermatomiositis y en los países más alejados la polimiositis era la entidad más frecuente. Estas diferencias se atribuyeron al influjo de la radiación UV como estímulo etiopatogénico. Estos resultados se han confirmado en otro estudio54,55. Otra teoría que no ha acabado de consolidarse hace referencia al fenómeno del microquimerismo fetal mediante el cual se cree que células inmunocompetentes del feto quedan anidadas en el seno materno y se activan produciendo una auténtica reacción de injerto contra huesped56,57. Aunque estudios epidemiológicos no han sido capaces de adscribir a las prótesis de silicona papel alguno en la etiopatogenia de las enfermedades autoinmunitarias en general y tampoco en las miositis tipo polimiositis o dermatomiositis, se han descrito casos de dermatomiositis en relación con prótesis de silicona en el contexto de un sustrato genético favorable58–60.
El endotelio capilar es el principal lugar de ataque en las dermatomiositis, y el complemento en su fracción C5b9 o complejo de ataque de membrana, la principal causa de la lesión capilar. Citocinas, moléculas de adhesión vascular e intersticial y metaloproteasas parecen desempeñar un papel adyuvante en el proceso inflamatorio liderado por células T y B. El mecanismo lesional parece distinto en la polimiositis donde la citotoxicidad mediada por linfocitos T CD8+ parece ser la causa principal a través de las perforinas.
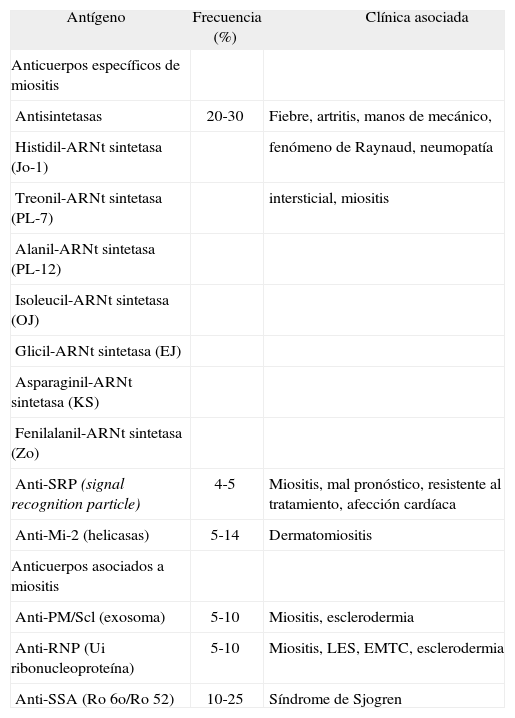
La presencia de autoanticuerpos es una de las características de las enfermedades autoinmunitarias. En la dermatomiositis y la polimiositis más de la mitad de los pacientes presentan anticuerpos antinucleares positivos y hasta un 20%, anticuerpos específicos o asociados a la miositis47,61–64. De forma mucho menos frecuente se ha descrito la presencia de estos autoanticuerpos en la MCI65,66. En la tabla 5 se especifican los principales anticuerpos relacionados con las miositis.
Autoanticuerpos en polimiositis/dermatomiositis
| Antígeno | Frecuencia (%) | Clínica asociada |
| Anticuerpos específicos de miositis | ||
| Antisintetasas | 20-30 | Fiebre, artritis, manos de mecánico, |
| Histidil-ARNt sintetasa (Jo-1) | fenómeno de Raynaud, neumopatía | |
| Treonil-ARNt sintetasa (PL-7) | intersticial, miositis | |
| Alanil-ARNt sintetasa (PL-12) | ||
| Isoleucil-ARNt sintetasa (OJ) | ||
| Glicil-ARNt sintetasa (EJ) | ||
| Asparaginil-ARNt sintetasa (KS) | ||
| Fenilalanil-ARNt sintetasa (Zo) | ||
| Anti-SRP (signal recognition particle) | 4-5 | Miositis, mal pronóstico, resistente al tratamiento, afección cardíaca |
| Anti-Mi-2 (helicasas) | 5-14 | Dermatomiositis |
| Anticuerpos asociados a miositis | ||
| Anti-PM/Scl (exosoma) | 5-10 | Miositis, esclerodermia |
| Anti-RNP (Ui ribonucleoproteína) | 5-10 | Miositis, LES, EMTC, esclerodermia |
| Anti-SSA (Ro 6o/Ro 52) | 10-25 | Síndrome de Sjogren |
EMTC: enfermedad mixta del tejido conectivo. LES: lupus eritematoso sistémico.
Diversos estudios epidemiológicos establecen sin lugar a dudas una estrecha relación entre dermatomiositis y cáncer, y en menor medida también entre polimiositis y cáncer67–69. Una tercera parte de los pacientes con dermatomiositis tendrá cáncer o, lo que es lo mismo, un paciente con dermatomiositis tiene un riesgo 3 veces más alto de desarrollar cáncer que un sujeto sin la enfermedad. Estos valores son algo más moderados para la polimiositis. A día de hoy no parece haber suficiente evidencia que permita asociar cáncer con MCI, lo que representa otro hecho diferencial de esta entidad. Entre las neoplasias que se asocian con mayor frecuencia destaca el carcinoma de ovario, por lo que algún estudio recomienda la determinación de marcadores tumorales de esta neoplasia anualmente durante los primeros 5 años del diagnóstico70,71. La representación de cáncer asociado a dermatomiositis, al margen de la neoplasia de ovario, parece seguir la normal distribución de cáncer de la población.
En general, el cáncer asociado a dermatomiositis-polimiositis tiene un comportamiento paraneoplásico; aparece 2–3 años antes o después del diagnóstico de miopatía inflamatoria. Aunque se acepta que el curso de la dermatomiositis y la polimiositis corre paralelo al del cáncer, es difícil de saber, ya que la mejoría de aquellas tras el tratamiento quimioterápico puede deberse a la resolución de la neoplasia o bien a la potente acción inmunodepresora del tratamiento antineoplásico.
Se recomienda al inicio del diagnóstico de dermatomiositis-polimiositis un cribado que incluya una analítica general con perfil básico de sangre y bioquímica, marcadores tumorales de próstata, ovario y mama, TC torácica y abdominal y mamografía y exploración ginecológica. Aun así, no es infrecuente que el seguimiento clínico del paciente nos sorprenda con la aparición de una neoplasia, por lo que se han desarrollado nuevas estrategias72. La tomografía por emisión de positrones (PET)-TC con fluorodesoxiglucosa, técnica híbrida que une morfología y funcionalismo celular, podría ser una buena opción, aunque resultados preliminares (observación personal, datos no publicados) no demuestran una mayor rentabilidad que el cribado convencional; de todas maneras hacen falta estudios más amplios para establecer su utilidad en este aspecto.
Varios estudios han puesto de manifiesto la existencia de antígenos compartidos por células tumorales y miocitos73–75. El reciente descubrimiento de anticuerpos frente a la proteína de 155 kD parece tener un elevado poder predictivo negativo, es decir, los pacientes con resultado negativo para este autoanticuerpo es improbable que desarrollen cáncer76–78.
TratamientoEl tratamiento de las miopatías inflamatorias se basa en la administración de glucocorticoides e inmunosupresores, sin olvidar la terapia física o de rehabilitación, incluso en la fase aguda79,80. Una tercera parte de los pacientes responde al tratamiento único con glucocorticoides, pero la mayoría precisa de la adición de un nuevo inmunosupresor. Entre éstos, azatioprina81 a dosis de 1–2mg/kg/día ajustada según el resultado del polimorfismo de la tiopurina metiltransferasa y metotrexato82,83 en dosis de 7,5-20mg/semana son los más conocidos. Sin embargo, los antagonistas de la calcineurina, como la ciclosporina o el tacrolimus, cuando el paciente no es hipertenso, son bien tolerados y especialmente útiles en la afección intersticial83–85. También la administración de ciclofosfamida en pulsos mensuales de aproximadamente 700mg junto con mesna para disminuir la toxicidad vesical, durante 6 meses, ha demostrado su utilidad en el control de la neumonitis intersticial en estos pacientes86. Nuevos inmunosupresores con acción contra el metabolito, como el micofenolato mofetilo o el ácido micofenólico, han demostrado una potencial utilidad en el tratamiento de las miositis resistentes87, si bien se han descrito ya casos de procesos linfoproliferativos en relación con su administración. Las inmunoglobulinas intravenosas88,89 actúan mejorando la debilidad muscular, son rápidas en su efecto, poco tóxicas y bien toleradas, el problema es su precio y que actúan de forma sintomática, en cualquier caso, pueden utilizarse de puente hasta conseguir el efecto esperado con el tratamiento inmunosupresor. La plasmaféresis, la irradiación corporal total o la ciclofosfamida oral no han demostrado eficacia en el tratamiento de las miopatías inflamatorias90,91. Otros fármacos menores, como los antipalúdicos de síntesis, útiles en otras enfermedades sistémicas como, por ejemplo, el lupus, podrían utilizarse como fármaco adyuvante en el control de las manifestaciones cutáneas y/o articulares, aunque nuestra experiencia es mala en este sentido. También se ha descrito la utilidad de suplementos de creatinina como terapia coadyuvante92. Múltiples ensayos y tratamientos utilizados en pacientes con MCI han fracasado o no han sido concluyentes93–97. Las terapias biológicas, como etanercept, infliximab o rituximab, han mostrado eficacia en algunos casos clínicos o estudios observacionales con pocos pacientes98,99. Actualmente está en marcha un estudio aleatorizado, a doble ciego, con placebo y rituximab, del que se esperan los resultados.


