

To evaluate the effectiveness and safety of tocilizumab (TCZ) in patients with rheumatoid arthritis (RA) in clinical practice, establishing the optimised regimen and switching from intravenous (IV) to subcutaneous (SC) therapy.
Material and methodsRetrospective observational study. We included 53 RA patients treated with TCZ. The main outcome was TCZ effectiveness at week 24. Secondary outcome variables included effectiveness at week 52, therapeutic maintenance, physical function and safety. The effectiveness of optimization and the switch from IV to SC was evaluated at 3 and 6 months. The efficacy was measured with the Disease Activity Score. Paired t-tests or Wilcoxon were used to evaluate effectiveness and survival time using Kaplan–Meier.
ResultsThe proportion of patients who achieved remission or low disease activity at weeks 24 and 52 was 75.5% and 87.3%, respectively. The mean retention time (95% confidence interval [95% CI] was 81.7 months [76.6–86.7]). Twenty-one of 53 patients (39.6%) optimised the TCZ dose and 35 patients switched from IV TCZ to SC, with no changes in effectiveness. The adverse event rate was 13.6 events/100 patient-years.
ConclusionsTocilizumab appears to be effective and safe in RA in clinical practice. The optimised regimen appears to be effective in most patients in remission, even when they change from IV to SC.
Evaluar la efectividad y la seguridad de tocilizumab (TCZ) en pacientes con artritis reumatoide (AR) en práctica clínica; la optimización de dosis y el cambio de formulación intravenosa (iv) a subcutánea (sc).
Material y métodosEstudio observacional retrospectivo. Se incluyó a 53 pacientes con AR tratados con TCZ. El desenlace principal fue efectividad de TCZ en la semana 24. Variables de desenlace secundarias incluyeron: efectividad en la semana 52, tiempo de retención del tratamiento, función física y seguridad. También se midió efectividad de la optimización de dosis y del cambio de formulación iv a sc a los 3 y 6 meses. La efectividad se midió con el índice de actividad según el Disease activity score-28. Se usó la prueba T pareada o prueba de rangos con signos de Wilcoxon para evaluar efectividad y el tiempo de supervivencia mediante curvas de Kaplan-Meier.
ResultadosLa proporción de pacientes que alcanzaron la remisión o baja actividad de la enfermedad en las semanas 24 y 52 fue del 75,5 y el 87,3%, respectivamente. La media de tiempo de retención (intervalo de confianza del 95% [IC del 95%]) fue de 81,7 meses (76,6-86,7). Veintiuno de 53 pacientes (39,6%) optimizaron la dosis de TCZ y 35 pacientes cambiaron a TCZ sc desde iv, sin cambios en resultados de efectividad. La tasa de efectos adversos fue 13,6 eventos/100 pacientes-año.
ConclusionesTocilizumab parece efectivo y seguro en AR en práctica clínica. La reducción de dosis parece efectiva en la mayoría de los pacientes en remisión, incluso cuando cambian de iv a sc.
Rheumatoid arthritis (RA) is a chronic inflammatory disease which without treatment may lead to joint destruction and functional damage.1 Although conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs) have been the foundation of therapy for many years, use of biologics has been associated with higher rates of remission in RA.2
Although biologic therapies have been a major advance in the control of RA, their potential toxicity and high cost3,4 must be taken into consideration. This has led to the question about what to do with patients whose status is sustained remission. One fairly well-used strategy has been dose reduction and even its discontinuity. However, in several studies 40%–75% of patients with sustained remission relapse after discontinuity of treatment with TNF antagonists.5,6 For this reason discontinuity of biologic treatment is no longer recommended. However, several studies indicate that a good percentage of patients may maintain remission with lower doses of biologics than those approved in the product data sheet.4,7
Tocilizumab is a humanised monoclonal antibody targeted against the soluble, membrane interleucine-6 receptor.8 Phase III clinical trials involving tocilizumab have demonstrated efficacy and safety in patients with RA who have presented with an inappropriate response to methotrexate or to other sc csDMARDs or TNF-alpha inhibitors.9–11 Although tocilizumab was initially approved for intravenous (iv) use,12 subsequent approval of a subcutaneous (sc) formula has entailed lower costs and an improved quality of life for the patient.13 In one phase III double-blind multicentre clinical trial (MUSASHI) the non inferiority of sc tocilizumab at a dose of 162mg every 2 weeks compared with 8mg/kg iv every 4 weeks13 was confirmed. Also, another 2 international phase III trials, SUMMACTA and BREVACTA,14,15 verified not just the non inferiority of subcutaneous tocilizumab compared with iv, but also the superiority of sc tocilizumab compared with placebo.
In this study we aimed to assess the effectiveness and safety of tocilizumab in patients with RA for a period of up to 52 weeks under clinical practice conditions, and also the optimization of doses and change in formula from iv to sc.
Patients and MethodsDesign and Scope of the StudyRetrospective observational study based on the review of the clinical files of RA patients who had been treated with tocilizumab in the University Regional Hospital of Malaga, Spain. The study was approved by the Clinical Research Ethics Committee of the hospital (CEIC).
PatientsAll the patients treated with tocilizumab between January 2009 and December 2015 were recruited in the study. The patients were assessed on initiating treatment with tocilizumab and after 24 and 52 weeks of follow-up. The eligibility criteria were: age ≥18 years, RA according to the criteria of the American College of Rheumatology/European League against Rheumatism 201016 and having received an iv infusion with tocilizumab. Patients with any type of inflammatory or rheumatic disease which was not RA were excluded (except for secondary Sjögren syndrome).
Study ProtocolAll patients were normally prospectively followed up in a specific biologic therapy unit (BTU) in compliance with the pre-established protocol of systematic data collection. This protocol includes, among other variables, data on the disease activity, physical function and adverse effects. The BTU reviewed the patients treated with sc bDMARDs every 3 months in general and specific (of sc biologics) consultations, alternating between them. The patients with iv bDMARDs were reviewed every time the drug was administered. Five days after the 1st infusion or in the 2nd dose of the sc bDMARD the patients received a call from the nursing staff to confirm that everything had gone well. The patients initially received 8mg/kg tocilizumab every 4 weeks. During the treatment period with tocilizumab, there were no restrictions in the concomitant use of the DMARDs (i.e. methotrexate or leflunomide). The tocilizumab dose was optimised in several patients from 8 to 4mg/kg per infusion. The decision regarding the dose or intervals was initially based on the product data sheet but was changed in accordance with the practitioner's criteria. These changes also included the change from iv to sc administration, when the latter was available.
Measurements and VariablesThe main outcome was the effectiveness of tocilizumab at week 24. The secondary outcome variables included: effectiveness at week 52, treatment retention time, physical function and safety. Effectiveness of dose optimization was also measured, and the change of formulation from iv to sc after 3 and 6 months.
The effectiveness of tocilizumab was measured by using the proportion of patients with a low disease activity, measured by the disease activity score in 28 joints with erythrocyte sedimentation rate (DAS28-ESR<3.2) and remission (DAS28-ESR<2.6)17 at weeks 24 and 52. The following were also used as measurements of effectiveness: the retention rate of tocilizumab (i.e. the proportion of patients whose biologic treatment was not discontinued) at week 52 and physical function, measured with the health assessment questionnaire (HAQ) (range 0–3)18 at week 24 and 52. Safety was assessed by the total and serious adverse effects incidence rate between 2009 and 2015. This was conducted by dividing the total number of adverse effects by the sum follow-up time of all patients in years (number of events/patients–year). The serious adverse effects were defined as life-threatening or resulting in death, hospitalisation or long term disability. Serious infections were defined as requiring iv antibiotics or leading to hospitalisation or prolonged hospital stay or death. The effectiveness of optimization and change of iv route to sc route in treatment with tocilizumab was evaluated using the differences in DAS28-ESR, HAQ, C-reactive protein (CRP) and ESR at 3 and 6 months compared with the baseline value.
Other recorded variables include age (years), gender, weight (kg), height (cm), body mass index (BMI=weight/height2) and smoker habits (smoker, non smoker). RA variables included duration of symptoms, number of swollen joints (28 joints), number of painful joints (28 joints) and general assessment by the patient of their activity (measured on a visual analogue scale of 0–100mm). High, moderate and low activity, and also remission were identified as scores DAS28-ESR15 of >5.1, 3.2–5.1, 2.6–3.2 and <2.6, respectively. Concomitant use of DMARDs was recorded.
Statistical AnalysisDescriptive analysis was made expressing the quantitative variables as measurements of central and dispersion tendencies and qualitative variables as absolute and relative frequencies. The Kolgomorov–Smirnov test was used to confirm the fit to normality of continuous variables. The Student's t-test was used for independent samples or the Mann–Whitney test in cases of non normality between the DAS28-ESR, HAQ, CRP, ESR mean at 24 and 52 weeks. It was also used for the study of dose optimization and the change of formula at 12 and 24 weeks compared with baseline. The survival time of tocilizumab was analysed using the Kaplan–Meier curves. Survival probabilities and their 95% confidence interval (95% CI) were calculated. For all analysis a value of P<.05 was considered significant. Data were analysed with the IBM SPSS Statistics version 19 software.
ResultsBetween January 2008 and December 2015 (both inclusive), 55 patients with RA were treated in our service. Two patients were excluded because data was lacking on the main variables. Finally, a total of 53 patients were recruited, 51 began with iv tocilizumab iv and 2 with sc tocilizumab. Of these 21 (39.6%) had their initial dose of tocilizumab reduced and another 35 (66%) changed from iv to sc. Eighteen patients (34%) began treatment with tocilizumab in a clinical trial and later were administered the commercially named drug.
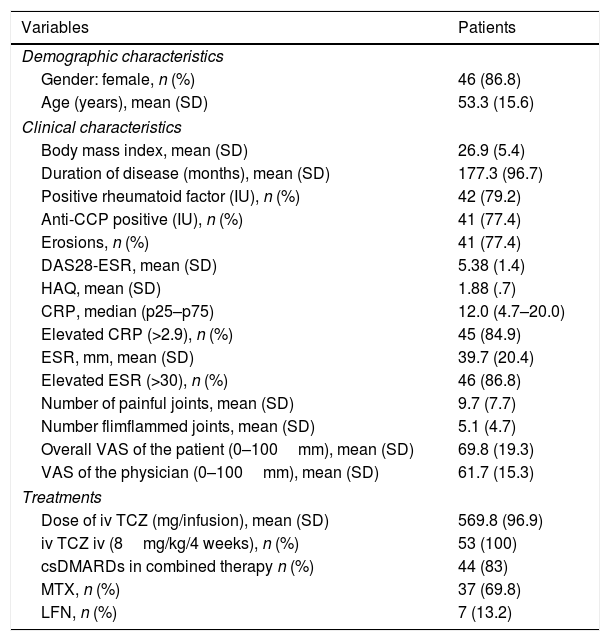
Patient CharacteristicsThe main patient characteristics are described in Table 1. The majority were women around 53 years of age and with a seropositive disease, which was erosive and long term. The great majority had high inflammatory activity of the disease at the beginning of treatment. Mean exposure (SD) to tocilizumab was 44.8 (19.4) months. The majority received tocilizumab in combination with a DMARD.
Baseline Characteristics of 53 Patients With RA Treated With Tocilizumab.
| Variables | Patients |
|---|---|
| Demographic characteristics | |
| Gender: female, n (%) | 46 (86.8) |
| Age (years), mean (SD) | 53.3 (15.6) |
| Clinical characteristics | |
| Body mass index, mean (SD) | 26.9 (5.4) |
| Duration of disease (months), mean (SD) | 177.3 (96.7) |
| Positive rheumatoid factor (IU), n (%) | 42 (79.2) |
| Anti-CCP positive (IU), n (%) | 41 (77.4) |
| Erosions, n (%) | 41 (77.4) |
| DAS28-ESR, mean (SD) | 5.38 (1.4) |
| HAQ, mean (SD) | 1.88 (.7) |
| CRP, median (p25–p75) | 12.0 (4.7–20.0) |
| Elevated CRP (>2.9), n (%) | 45 (84.9) |
| ESR, mm, mean (SD) | 39.7 (20.4) |
| Elevated ESR (>30), n (%) | 46 (86.8) |
| Number of painful joints, mean (SD) | 9.7 (7.7) |
| Number flimflammed joints, mean (SD) | 5.1 (4.7) |
| Overall VAS of the patient (0–100mm), mean (SD) | 69.8 (19.3) |
| VAS of the physician (0–100mm), mean (SD) | 61.7 (15.3) |
| Treatments | |
| Dose of iv TCZ (mg/infusion), mean (SD) | 569.8 (96.9) |
| iv TCZ iv (8mg/kg/4 weeks), n (%) | 53 (100) |
| csDMARDs in combined therapy n (%) | 44 (83) |
| MTX, n (%) | 37 (69.8) |
| LFN, n (%) | 7 (13.2) |
Anti-CCP: anti-cyclic citrullinated peptide antibodies; DAS28: disease activity score on 28 joints; VAS: visual analogue scale; csDMARDs: conventional synthetic disease-modifying anti-rheumatic drugs; HAQ: Health Assessment Questionnaire; LFN: leflunomide; MTX: methotrexate; CRP: C-reactive protein; TCZ: tocilizumab; ESR: erythrocyte sedimentation rate.
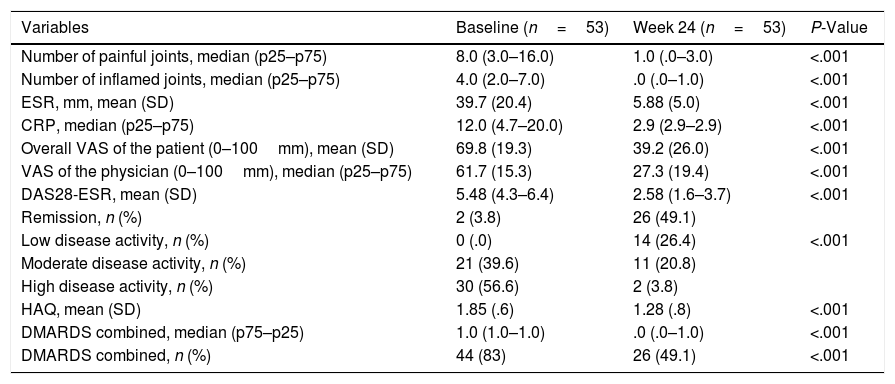
Out of the 53 patients treated with tocilizumab, 51 (96.2%) continued receiving tocilizumab in week 24 and 52. None of the patients discontinued tocilizumab due to insufficient response during the observation period at 52 weeks. As shown in Table 2, the patients treated with tocilizumab had substantially improved by week 24. The proportion of patients who achieved remission or low activity of the disease in weeks 24 and 52 was 75.5% and 87.3%, respectively. Furthermore, patients also demonstrated better disease control at week 52, measured by DAS28-ESR, ESR, CRP and HAQ. Mean retention time (95% CI) was 81.7 months (76.6–86.7). Patients with moderate to high disease activity in week 52 were associated with a better BMI compared with patients with low activity/remission (25.6 vs 28.9; P=.02).
Differences in Clinical Characteristics Between Baseline and Week 24.
| Variables | Baseline (n=53) | Week 24 (n=53) | P-Value |
|---|---|---|---|
| Number of painful joints, median (p25–p75) | 8.0 (3.0–16.0) | 1.0 (.0–3.0) | <.001 |
| Number of inflamed joints, median (p25–p75) | 4.0 (2.0–7.0) | .0 (.0–1.0) | <.001 |
| ESR, mm, mean (SD) | 39.7 (20.4) | 5.88 (5.0) | <.001 |
| CRP, median (p25–p75) | 12.0 (4.7–20.0) | 2.9 (2.9–2.9) | <.001 |
| Overall VAS of the patient (0–100mm), mean (SD) | 69.8 (19.3) | 39.2 (26.0) | <.001 |
| VAS of the physician (0–100mm), median (p25–p75) | 61.7 (15.3) | 27.3 (19.4) | <.001 |
| DAS28-ESR, mean (SD) | 5.48 (4.3–6.4) | 2.58 (1.6–3.7) | <.001 |
| Remission, n (%) | 2 (3.8) | 26 (49.1) | |
| Low disease activity, n (%) | 0 (.0) | 14 (26.4) | <.001 |
| Moderate disease activity, n (%) | 21 (39.6) | 11 (20.8) | |
| High disease activity, n (%) | 30 (56.6) | 2 (3.8) | |
| HAQ, mean (SD) | 1.85 (.6) | 1.28 (.8) | <.001 |
| DMARDS combined, median (p75–p25) | 1.0 (1.0–1.0) | .0 (.0–1.0) | <.001 |
| DMARDS combined, n (%) | 44 (83) | 26 (49.1) | <.001 |
DAS28: disease activity score on 28 joints; VAS: visual analogue scale; csDMARDs: conventional synthetic disease-modifying anti-rheumatic drugs; HAQ: Health Assessment Questionnaire; LFN: leflunomide; MTX: methotrexate; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate.
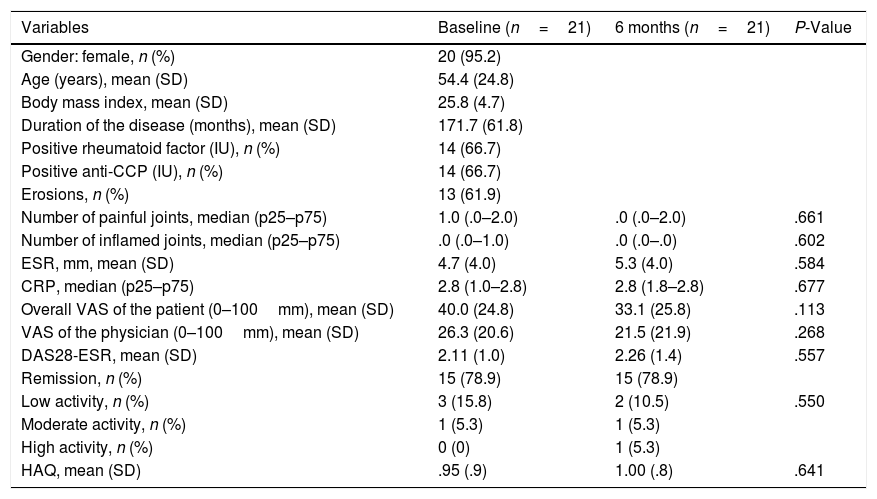
Twenty one of the 53 patients (39.6%) optimised the tocilizumab dose, 19 with iv and 2 with sc. All the patients in iv treatment reduced the dose of each infusion and maintained the interval between doses, whilst all patients with sc tocilizumab extended the interval between doses and maintained the dose. Two patients who had reduced doses with iv tocilizumab changed directly to sc with the reduced dose. The main baseline characteristics of the patients who reduced the doses are listed in Table 3. After 6 months, no differences were observed in efficacy results (Table 3), patient in remission percentage and low baseline activity of the disease and at 6 months it was situated at 94.7% and 89.4% respectively. Only one patient regressed to a complete dose of tocilizumab after 37 months of reduced dose due to clinical worsening of symptoms. No adverse effects were observed during follow-up.
Epidemiological and Clinical Characteristics of 21 Patients With RA Who Optimised Treatment With Tocilizumab.
| Variables | Baseline (n=21) | 6 months (n=21) | P-Value |
|---|---|---|---|
| Gender: female, n (%) | 20 (95.2) | ||
| Age (years), mean (SD) | 54.4 (24.8) | ||
| Body mass index, mean (SD) | 25.8 (4.7) | ||
| Duration of the disease (months), mean (SD) | 171.7 (61.8) | ||
| Positive rheumatoid factor (IU), n (%) | 14 (66.7) | ||
| Positive anti-CCP (IU), n (%) | 14 (66.7) | ||
| Erosions, n (%) | 13 (61.9) | ||
| Number of painful joints, median (p25–p75) | 1.0 (.0–2.0) | .0 (.0–2.0) | .661 |
| Number of inflamed joints, median (p25–p75) | .0 (.0–1.0) | .0 (.0–.0) | .602 |
| ESR, mm, mean (SD) | 4.7 (4.0) | 5.3 (4.0) | .584 |
| CRP, median (p25–p75) | 2.8 (1.0–2.8) | 2.8 (1.8–2.8) | .677 |
| Overall VAS of the patient (0–100mm), mean (SD) | 40.0 (24.8) | 33.1 (25.8) | .113 |
| VAS of the physician (0–100mm), mean (SD) | 26.3 (20.6) | 21.5 (21.9) | .268 |
| DAS28-ESR, mean (SD) | 2.11 (1.0) | 2.26 (1.4) | .557 |
| Remission, n (%) | 15 (78.9) | 15 (78.9) | |
| Low activity, n (%) | 3 (15.8) | 2 (10.5) | .550 |
| Moderate activity, n (%) | 1 (5.3) | 1 (5.3) | |
| High activity, n (%) | 0 (0) | 1 (5.3) | |
| HAQ, mean (SD) | .95 (.9) | 1.00 (.8) | .641 |
Anti-CCP: anti-cyclic citrullinated peptide antibodies; DAS28: disease activity score on 28 joints; VAS: visual analogue scale; HAQ: Health Assessment Questionnaire; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate.
Thirty five patients changed to sc formulation from iv and depending on the previous dose of iv tocilizumab (i.e. 8mg/kg, 6mg/kg or 4mg/kg) which was being received they were prescribed with the following sc tocilizumab dose: 162mg/week (n=30), 162mg/10 days (n=4) or 162mg/15 days (n=1), respectively. At 6 months, no differences were observed in the efficacy results when efficacy of sc formulation was compared with iv formation. The percentage of patients in remission and low baseline disease activity at 6 months was 88.6% and 77.3%, respectively. Four (11.4%) patients was readministered iv tocilizumab due to subjective worsening of symptoms (increase in the number of painful joints and assessment by the patients, without any increase in acute phase reactant tests, n=2) or for non-serious side effects (n=2).
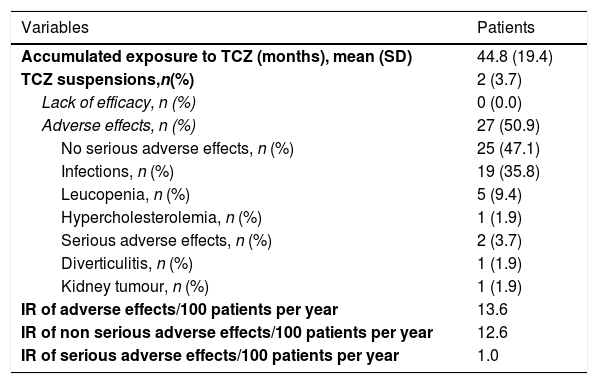
Safety of TocilizumabAs shown in Table 4, half of the patients presented with an adverse effect (13.6 events/100 patients per year), but only 2 of them were serious (1.0 case/100 patients per year), which required treatment withdrawal. The majority of side effects were infections. During treatment with sc tocilizumab, 12 (34.3%, .55 events/100 patients per year) patients had non serious adverse effects: 8 injection site reactions (22.8), 2 respiratory infections (5.71), one patient presented with hypertransaminasemia (2.85) and one patient presented with a vasovagal reaction (2.85). There were no serious adverse effects. There were no differences regarding the number of adverse effects between iv and sc tocilizumab (15 vs 12, P=.158).
Causes of Suspension and Adverse Effects of Tocilizumab.
| Variables | Patients |
|---|---|
| Accumulated exposure to TCZ (months), mean (SD) | 44.8 (19.4) |
| TCZ suspensions,n(%) | 2 (3.7) |
| Lack of efficacy, n (%) | 0 (0.0) |
| Adverse effects, n (%) | 27 (50.9) |
| No serious adverse effects, n (%) | 25 (47.1) |
| Infections, n (%) | 19 (35.8) |
| Leucopenia, n (%) | 5 (9.4) |
| Hypercholesterolemia, n (%) | 1 (1.9) |
| Serious adverse effects, n (%) | 2 (3.7) |
| Diverticulitis, n (%) | 1 (1.9) |
| Kidney tumour, n (%) | 1 (1.9) |
| IR of adverse effects/100 patients per year | 13.6 |
| IR of non serious adverse effects/100 patients per year | 12.6 |
| IR of serious adverse effects/100 patients per year | 1.0 |
IR: incidence rate; TCZ: tocilizumab; 100PY: 100 patients per year.
This is a descriptive study of our clinical practice with tocilizumab in the treatment of RA. According to our results, tocilizumab obtained good control of the clinical activity in practically all patients at 24 weeks and remained stable up to week 52. Moreover, we found an association between inflammatory activity measured by DAS28-ESR and BMI at week 52 of treatment. This has already been reported in the MUSASHI study20 and indicates that the efficacy of a fixed dose tocilizumab regimen may be lower in patients with high body weight. These results are therefore similar to those observed in other studies, both observational12,21 and experimental,10,14,15,19,22,23 but our study provides several novelties which have not been previously analysed in clinical practice, such as the measurement of DAS28-ESR remission not just at 24 weeks, as previously described,21 but also at 52 weeks, in addition to the transition between the 2 formulations (i.e. from iv to sc) and the lowering of dosing during this transition.
Results indicate that the tocilizumab dose may be reduced in patients in clinical remission after at least 6 months without losing its effectiveness. Only one patient regressed to the complete dose after 37 months due to worsening of clinical symptoms. Cases of increased activity have also been reported in other studies after the reduction of treatment with iv tocilizumab,4,15 and was justified by the special influence this drug has on the acute phase reactants and therefore on the level of DAS28-ESR activity. In this regard, it is of note that the sustained reduction of inflammation markers with a 8mg/kg dose of tocilizumab, but not with a 4mg/kg dose of tocilizumab has been reported.4,24
We also observed that efficacy was appropriately maintained in the majority of patients who changed formulation. The reduced iv dose could also be maintained in sc administration without the need to return to an upper standard sc dose. The results from the MUSASHI study indicate that the majority of patients may be changed from iv to sc tocilizumab with no reduction in efficacy or safety concerns.25 However, 2 of our patients regressed to iv tocilizumab due to subjective worsening of symptoms and probably due to a nocebo effect.
We did not observe any particular safety problems with tocilizumab in our study. For the 52 weeks 12.6 non serious adverse events were observed per 100 patients per year. As expected, the frequency of reactions at the injection site with the sc formulation was slightly higher at .36 local reactions per 100 patients per year.26 All reactions at the injection site were mild and in no case was treatment discontinued. Respiratory infections were the most common (5.7 episodes/100 patients per year), which also replicated results from other observational studies.12 Two serious adverse effects forced us to discontinue treatment: one diverticulitis and one kidney tumour. A systematic review and clinical trial meta-analysis found that adverse effects increased in patients who received tocilizumab combined with methotrexate compared with the monotherapy (odds ratio [OR] 1.5, 95% CI, 1.3–1.9), as was the rate of infection (OR 1.3, 95% CI, 1.1–1.6).19 Notwithstanding, there was no significant increase in malignancy rates, reactivation of tuberculosis or hepatitis.27,28
Our study has several limitations, including its retrospective design and relatively small sample size. Although design is technically retrospective, it is actually mostly prospective, since all variables analysed had been collected prospectively and systematically, based on previously designed protocol. This explains the zero rate of data loss, with only 2 patients excluded, and such consistent results. The number of patients treated with tocilizumab is low because data are from a single hospital and this had a particularly negative impact for sub analysis of dose reduction in changing formulation from iv to sc. In our study the patients whose dose was reduced were in remission or disease activity was low prior to reduction, where it was less probable that the patients’ condition would worsen after being stable for some time. As described in other studies, this persistent effectiveness could be due to prior control of the disease.29 For this reason, concern continues for these patients in case they experience outbreaks of the disease long term which could cause radiologic damage and which has not been analysed in our study.4,30 We could resolve this limitation in posterior studies with prospective follow-up in these patients with both inflammatory activity markers, and assessment of radiologic damage, together with laboratory analysis such as the rheumatoid factor and the citrullinated cyclic peptide factor. We would state, therefore, that 6 months is possibly an insufficient period to evaluate the efficacy of dose optimization and the change from iv to sc tocilizumab formulations in clinical practice conditions. This could be improved by following our cohort for a longer length of time.13
To conclude, these results highlight tocilizumab's effectiveness in safely controlling the signs and symptoms of RA patients in clinical practice. This control is maintained in the majority of patients in remission with lower doses than those of the product data sheet even when changing formulation from iv to sc. Confirmation of these results requires specific studies for a longer time period, but the possible benefits of this strategy would include cost reduction and fewer adverse effects, depending on the dose.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that for this research no experiments have been carried out on humans or animals.
Confidentiality of dataThe authors declare that they have adhered to the protocols of their centre of work on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appears in this article.
Conflict of InterestsThe authors have no conflict of interests to declare.
Please cite this article as: Mena-Vázquez N, Manrique-Arija S, Rojas-Giménez M, Ureña-Garnica I, Jiménez-Núñez FG, Fernández-Nebro A. Análisis de la efectividad, seguridad y optimización de tocilizumab en una cohorte de pacientes con artritis reumatoide en práctica clínica. Reumatol Clin. 2019;15:21–26.


