


There is no doubt that biologic therapies provide added value for health systems. However, due to their special nature, they also raise some questions that make highly rigorous and demanding quality control and monitoring of their use indispensable. This circumstance is reinforced with the appearance on the scene of biosimilars, which, given their lower cost, are having an increasing impact on the international market. The purpose of this article is to review the major issues posed by their manufacture, distribution and control systems, as well as the most important aspects related to their safety in clinical practice. In this report, we assess the pharmacovigilance of these products, with special attention to traceability, as a key tool to enable earlier detection of adverse events.
No cabe duda de que los productos biológicos aportan un valor añadido a los sistemas de salud, aunque también plantean grandes interrogantes debido a su especial naturaleza, lo que obliga a ser muy rigurosos y exigentes en su control de calidad y seguimiento. Este hecho se ha visto reforzado por la entrada en escena de los fármacos biosimilares, cuyo menor coste está permitiéndoles alcanzar un mayor protagonismo en el mercado mundial. El propósito de este artículo es revisar en profundidad los principales interrogantes que se plantean en su producción, distribución y control, así como los aspectos más importantes relacionados con su seguridad en la práctica clínica. En este trabajo revisamos lo que representa la farmacovigilancia de estos productos, prestando especial atención a su trazabilidad, como herramienta fundamental para la detección precoz de acontecimientos adversos.
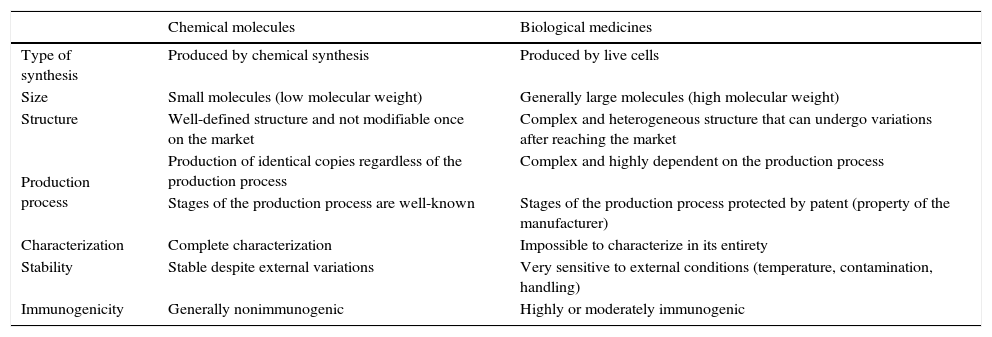
Biological agents represent a great advance in the treatment of complex diseases, such as rheumatoid arthritis (RA), psoriasis, Crohn's disease, multiple sclerosis, diabetes and cancer,1–4 for which, until they were introduced, we had no really efficient therapeutic options.5 However, their marked complexity in comparison with drugs obtained by traditional chemical synthesis (Table 1) means that their use requires special care.6 For this reason, they are included in the list of “medicines under additional monitoring” drawn up for the European Union (EU) pharmacovigilance system.7 Moreover, we have come to the moment when the patents of some of them are expiring and, with the arrival on the scene of biosimilars, there are a number of questions that we will attempt to address in this review.
Differences Between Chemically Synthesized Drugs and Biological Agents.
| Chemical molecules | Biological medicines | |
|---|---|---|
| Type of synthesis | Produced by chemical synthesis | Produced by live cells |
| Size | Small molecules (low molecular weight) | Generally large molecules (high molecular weight) |
| Structure | Well-defined structure and not modifiable once on the market | Complex and heterogeneous structure that can undergo variations after reaching the market |
| Production process | Production of identical copies regardless of the production process | Complex and highly dependent on the production process |
| Stages of the production process are well-known | Stages of the production process protected by patent (property of the manufacturer) | |
| Characterization | Complete characterization | Impossible to characterize in its entirety |
| Stability | Stable despite external variations | Very sensitive to external conditions (temperature, contamination, handling) |
| Immunogenicity | Generally nonimmunogenic | Highly or moderately immunogenic |
The objective of pharmacovigilance systems is the identification, quantification, evaluation and prevention of the risks of the use of drugs once they have been made commercially available. Therefore, they are designed to make decisions that enable the maintenance in the market of medications in relation to their benefit-risk ratio, or withdraw them from use when this is not possible. It is an activity in which the responsibility is shared among all the agents implied in the use of the drug: holders of the marketing authorizations, health authorities, physicians, pharmacists, nurses and patients, as well as those who assess reports of suspected adverse reactions.8 For the purpose of defining the bases that establish a quality system, in 2001, the European Medicines Agency (EMA) created EudraVigilance, a network for data processing to evaluate adverse events (AE) reported on the part of regulatory agencies and pharmaceutical companies throughout Europe.
Legislation regarding pharmacovigilance is constantly evolving. On the other hand, the planning of risk management and related activities can vary from one country or region to another, making it difficult to adapt it to the legal framework of each. Thus, the EU created a “risk assessment system” that encompasses all of the activities, and is committed to characterizing and minimizing the possible risks related to a medication. According to it, anyone who applies for the authorization of a medicine is obliged to present a risk management plan (RMP), the purpose of which is to guarantee patient safety, and its update, if considered necessary, can be required at any time.9
In the case of biological products, due to their particular characteristics, all of these activities should be done with great meticulousness. In fact, as “a group of medicines under additional monitoring” they include all those biologic products authorized since 2011, a group that they can eventually abandon, like other drugs, once the necessary time has elapsed. More specifically, and because of the complex production process, slight variations in them can affect the final product, even in products with the same active ingredient. Some examples of this are AE associated with erythropoietin, as occurred in 2002,10,11 with differences reported in the risk of developing factor VIII inhibitors depending on whether second or third-generation products are employed12 or an increase in the incidence of thrombotic microangiopathy in patients treated with interferon beta preparations with small variations, as was observed with Rebif® and Avonex®. This makes it indispensable to have access to systems capable of detecting any potential distinction in the different biological products, especially for changes produced during manufacture, meaning it is necessary to know the exact batch of each product, as well as the brand name (Fig. 1).

Variability in the safety of biological products beyond the content of the active ingredient. ATC, anatomical therapeutic chemical; INN, international nonproprietary name. *The examples are provided for illustrative purposes. It should be remembered that Remsima and Inflectra are the same product. Modification from Vermeer et al.48
The introduction of the first biological products in clinical practice in Europe brought with it special pharmacovigilance measures, the efficacy of which has been weighed. However, the number has grown a great deal in recent years, mainly due to the large amount of innovative new molecules, but also as a consequence of the arrival on the scene of biosimilar products (Appendix B Supplementary table). This generated a controversial situation centered on the safety and efficacy profiles of the new biological agents, accompanied by continuous debates about them and even on the original products.13–16 This could all be avoided by guaranteeing compliance with the current legislation and providing proper training in their utilization for all those involved.
There are a number of methods of evaluating the safety of biological drugs, once they become commercially available. The most well-known among professionals is the spontaneous reporting system, based on conveying information on AE that arise during routine clinical practice. At the present time, nearly every country in the world has systems that enable reporting of AE both to health professionals and to the patients themselves.17 However, very few AE are reported and the quality of the alerts can vary, there being many cases in which we cannot relate a certain AE to a responsible drug without the fear of committing an error. In fact, voluntary reporting of events related to drugs on the part of the hospital staff not is entirely reliable and its level of efficacy differs a great deal from one center to another, as is shown by various studies.18,19 As an added difficulty, it occurs that, despite the fact that current legislation requires the traceability of the biological medicines utilized, the degree to which this is carried out varies depending on the health center being considered.20
This is all very much related to having access to the available information. Thus, for AE that develop over a relatively short time, the essential data can be obtained from the product packaging; but if this has been discarded, the quality of the report will depend on each specific situation. In some cases, for example, physicians may encounter an AE from a medication prescribed by another, and that they do not have access to all the necessary information to carry out a correct clinical decision. In other cases, the patient may not be able to differentiate AE produced from the drug and the symptoms of the disease itself.
Another resource employed includes the electronic databases of the health systems (medical databases and those that collect claims or records concerned with a medication).21 They have the advantage of ensuring the routine and systematic storage of clinical data. Their availability will depend on the level of development and proper functioning of pharmaceutical records.
Finally, we wish to point out the databases and records kept by the numerous medical associations and scientific societies, which are becoming increasingly important. These vary from small national registries to large international databases that work with multiple treatment arms and focus on a wide range of clinical settings. The majority of these registries are based on selected long-term cohorts that provide information on treatments, as well as on the clinical outcomes obtained, all performed over predefined time intervals. This is the case of the Spanish registry of AE of biological therapies in rheumatic and cutaneous diseases (BIOBADASER and BIOBADADERM, respectively), the British Society of Rheumatology Biologics Register (BSRBR)22,23 or the European PedNet Haemophilia Registry,24 to give just a few examples.
On the web page of the EMA we can find all the information referring to the Spanish Pharmacovigilance System, which integrates the activities carried out by health administrations with regard to this subject and is constituted by the Autonomic Pharmacovigilance Centers and the Division of Pharmacoepidemiology and Pharmacovigilance of the EMA and the Spanish Ministry of Health and Consumer Affairs, as well as by health care professionals.25
Traceability of Biological AgentsTraceability is a key aspect within the field of pharmacovigilance. We understand this to be the ability to reconstruct the history, route or application of a pharmaceutical product, identifying the origin of its components, the methods utilized and its distribution, as well as where it was stored after its production and throughout all of its “life cycle”. It consists of being able to identify the stages that a product has been subjected to within the process of being manufactured (manipulations, composition, machinery employed, temperature to which it was subjected, batch number, etc.), which are considered important and that can vary the final product for the consumer. A proper traceability system should be implemented by all of the individuals or legal entities that intervene in the chain of the production, marketing, distribution and dispensing of the medication in question (laboratories, suppliers, logistic operators, pharmacists, physicians and nurses).26
The manufacturer provides an element or device capable of storing a unique and unequivocal code on the packaging of the products to be sold to the public, that enables the identification of each unit of the medication as singular, which is essential for constructing a chain to monitor it. It is also essential to have access to a database in which it is possible to add all the data on the drug as it continues to become available and that can be accessed simply and rapidly.27
In 2013, the European authorities endorsed a law (Directive 2011/62) on falsified medicines for all of the agents in the supply chain, according to which, starting in 2016, it will be obligatory for each product to have an individual barcode which will be stored in the records of the manufacturer and subsequently be corroborated in the dispensing records. The system chosen to ensure the traceability of these products was Data Matrix, which ruled out other reading procedures, like radiofrequency, which were also analyzed. Data Matrix, which will not share space on the packaging with any other identifying code, will include information on the identification of the product, the serial number, country code, batch number and expiration date.28
Biological agents are dispensed through several distribution systems in which the information is collected by scanning the barcode on the outer packaging, which stores information that accompanies the brand name of the product, which is unique for each manufacturer and dose. This enables automatic storing of all of the associated information.29 However, a handwritten note should be made of the product batch each time the medication is dispensed, using barcodes that should be processed by computer which, at the present time, are not included. On those occasions in which there is no available computer system for dispensing, all of this information should be handwritten to be stored (since for many administrative activities it is necessary to provide the time at which the supply was being handled). In a recent study, it was concluded that the batch number was taken down for the majority of the biological agents administered in hospitals, in contrast to what occurs in retail pharmacies. This is mainly due to adherence to good practices in preparation and to the “Guidelines for Plasma-derived Medicinal Products” which requires taking note of the batches of all of the products derived from plasma or blood.30
In most cases, there is a space in the hospital designated for the administration of biological agents, because of the complex processes involved both in their preparation and in their delivery, as well as the necessary monitoring required once the treatment has been administered. The information collected on the medication will depend, among other things, on whether the records are taken down on paper or in electronic format, a proper relationship between the pharmacy and the administration, and the protocols in each hospital for gathering information according to the type of biological agent utilized. However, in the latter respect, there are certain products that require special conditions regardless of the internal protocols of the hospital, as is the case of blood products or that of infliximab and its biosimilars, for which European guidelines recommend that both the health care administrations and the patient take down the batch number and expiration date at the time the product is dispensed.31
Fortunately, there are ever more initiatives for improving traceability systems. France was the first country in the EU to include the requirement that obliges the insertion in the information of the Data Matrix.32 Other countries, like Italy, Greece and Belgium, are moving in the same direction.33 There are indications that future advances in technology will enable the automatic annotation of both the batch number and the expiration date of each product separately.34,35 The development of applications for cell phones for use when the patients self-administer the medication at home is even foreseeable.36
Challenges and Uncertainties of Biological TreatmentInterchangeabilityChoosing the proper biological therapy and determining whether it is necessary to replace one biological treatment with another involves making a delicate decision. This can occur because of inefficacy or due to an AE resulting from a previous treatment. The introduction of biosimilars brings with it the development of less expensive and equally efficient therapies, but it also creates doubts as to whether or not a given treatment should be “switched” for another.
In the absence of specific biomarkers for many diseases, it is very difficult to establish universal characteristics that unmistakably distinguish concepts such as “treatment failure” and “inadequate response”. Thus, in RA, rheumatologists in Europe and North America have reached a consensus with respect to the definition of “disease remission” that directly effects the management of the patient.37 In addition, AE associated with biological therapies, like infusion reactions or the complications derived from the complex administration systems often used with these products, can result in the frustration of the patient, who starts to consider a therapy to be ineffective before it could even have an effect. A study published in recent years, but conducted before the arrival on the scene of biosimilar drugs, revealed that a third of the patients included had undergone more than 1 switch in the type of biological therapy utilized in a period of only 15 months.38
Thus, to insure acceptable interchangeability, the data on the therapy must be collected continuously. However, basing our decisions on observational safety studies to identify significant differences in events that occur infrequently, such as infusion reactions or severe infections, does not seem to be very reliable. It must be taken into account that these products are also utilized in countries in which there is a high prevalence of severe infections, such as tuberculosis, which can bias the interpretation of observational data on safety in infectious events. Moreover, the period during which switching can be carried out must be very much taken into account as it can be a determining factor. Thus, in patients in whom the response to treatment is lost due to the development of anti-drug antibodies, the change should be done between treatments.39
Another aspect that must be kept in mind is that biological agents can present modifications that means they may utilize an independent pathway, both in their manufacture, formulation, primary conditioning or even the discovery of new therapeutic indications. This could create a scenario in which the differences that may result between the innovator batches or between the different biosimilars of the same original product can increase over time.40 Taking this into account when it comes to ensuring biosimilarity is a great challenge in achieving a switch with sufficient guarantees since, to date, we have little scientific evidence in this area. Thus, we consider that this novel scenario in the therapeutic role of biological agents stemming from the development of the biosimilars implicates the challenge of consolidating all that is known about these medications on the part of those involved in their management, with the commitment of not compromising the efficacy or the safety that they have shown to date.41
Immunogenicity and Its Involvement in Quality, Efficacy and SafetyOne of the major problems encountered with these products is that there can be differences with respect to the dynamics of antibody formation when compared with the reference product or innovator. For example, in the case of adalimumab, the production of antibodies has been related to an increase in the development of thromboembolic episodes.42 However, there is no evidence that repeated switching introduces this immunogenicity per se and, therefore, each case has to be studied individually.43 With infliximab, the majority of the antibodies that act do so by binding to the murine part, whereas antibodies against adalimumab do so by binding to the complementarity-determining region.42
The formation of antibodies against these medications may have different mechanisms: hypersensitivity reactions, cross-linking of Fc receptors (which can produce reactions at the injection site) or immediate autoimmune responses not related to antibody production, which occurs with nearly all of the monoclonal antibodies on the market. One exception is cetuximab, in which case, preexisting anti-galactose-α-1,3-galactose antibodies have been related to fatal infusion reactions in patients who were receiving it for the first time.42 For this reason, reactions of this type should not represent an obstacle when it comes time for switching, and manufacturers of biosimilars with this product are utilizing smaller amounts of “nonhuman” glycans. Thus, we can expect that there will be fewer infusion reactions, with maintenance of the same efficacy shown by the innovator. This creates a paradoxical situation since, there could be occasions when the biosimilar would be less immunogenic than the innovator product and, even so, a switch cannot be done because of the risk of an immunogenic response.
In any case, on the basis of the limited literature available to date, we have found that the risk of immunogenicity is relative low after 12 months of treatment with infliximab. Only 90% of the patients who developed sustained antibodies, did so within the first 12 months of treatment.44 The same occurs in the case of adalimumab.45
Finally, certain caution should be utilized in the management of these data, which can be employed in comparative and complementary studies. The variability in the results depending on the diagnostic method used should also be taken into account.44 That is, if the major incident observed after 12 months of treatment was a potential change in immunogenicity, to establish its clinical relevance, it would be necessary to have access to a central laboratory and to the correlation of the outcomes with the content of the medicine in the organism, response to treatment and incidence and severity of AE.
Nomenclature and LabelingGiven that their molecular individuality makes it difficult for there to be 2 exact copies of these drugs, biological products should have a nomenclature that unequivocally defines and identifies them. As indicated by the EMA,46 the labeling of the medication and, particularly, the “directions for use”, are key aspects when it comes to marketing the product within the EU and constitute the basis for the information for health professionals to ensure its effective and safe utilization. The directions for use must be continuously updated, reflecting any change that occurs during the “life cycle” of the product. However, in the specific case of biological agents, it is not clear what data should be included. In fact, in the last expert meeting concerning biosimilar products, several alternatives were presented, and none of them achieved the approval of a majority.47
The World Health Organization (WHO) established a nomenclature system for drugs, the “International Nonproprietary Name” (INN) which is indicative of the active ingredient and of the therapeutic group. It is recognized universally and facilitates the unequivocal identification of active ingredients.47
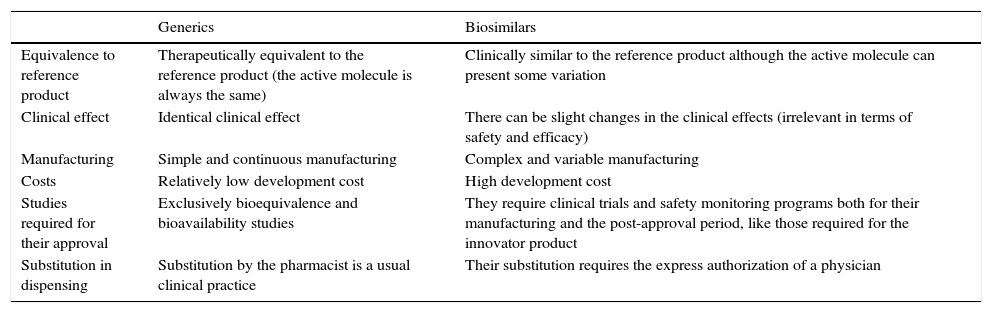
In the case of biosimilars, some of them generic, the medications should only be employed for the authorized indications, and, exceptionally, for others not considered among the directions for use when there are no other alternatives. However, biosimilar drugs, in contrast to generics, cannot be utilized automatically in all of the indications of the reference product, and any extrapolation of them requires a special justification (Table 2).
Differences Between Generics and Biosimilar Drugs and Their Corresponding Reference Products.
| Generics | Biosimilars | |
|---|---|---|
| Equivalence to reference product | Therapeutically equivalent to the reference product (the active molecule is always the same) | Clinically similar to the reference product although the active molecule can present some variation |
| Clinical effect | Identical clinical effect | There can be slight changes in the clinical effects (irrelevant in terms of safety and efficacy) |
| Manufacturing | Simple and continuous manufacturing | Complex and variable manufacturing |
| Costs | Relatively low development cost | High development cost |
| Studies required for their approval | Exclusively bioequivalence and bioavailability studies | They require clinical trials and safety monitoring programs both for their manufacturing and the post-approval period, like those required for the innovator product |
| Substitution in dispensing | Substitution by the pharmacist is a usual clinical practice | Their substitution requires the express authorization of a physician |
To avoid the error that this may produce when it comes to prescribing a biosimilar for an indication in which it has not been approved, the WHO has proposed to add a series of numbers and letters to the INN by means of prefixes and suffixes to classify molecules more accurately. Moreover, for biological drugs that have proteins with extensive glycosylated residues, it establishes the use of additional suffixes. In this way, the biologicals receive singular and “unique” names. This system has had a different degree of acceptance worldwide. In fact, the EMA, for now, has not decided to adopt it.47
However, despite these measures, the same name continues to be used for different products (with distinct manufacturers, processing, formulation, administration systems, etc.).48 One example is “interferon beta-1”. Recently, manufacturers of innovator products proposed to establish an additional INN system that would alert to the fact that a certain medication is a biosimilar. Therefore, manufacturers of biosimilars recall that, at the present time, innovators conserve the same INN although they present variations in their glycosylated residues, which is contradictory, as products approved as being highly similar have different names.49
Regulatory GuidelinesAt this point, we should remember that both the innovator product and the biosimilar are obliged to follow the procedures of a central registry coordinated by the EMA. The reference medication with which the biosimilar is being compared should be authorized in the EU and must be the same for the entire project for the development of the biosimilar. The EMA has groups of experts in biologics, such as the Biologics Working Party, and in biosimilars, in particular, the Biosimilar Medicinal Products Working Party, which share their opinions on all that has to do with these drugs for the use of the Committee for Medicinal Products for Human Use, which draws up the guidelines on biological medications, providing a scientific opinion on their approval. On this basis, the European Commission (EC) makes a final decision on their authorization and marketing in the EU.50 This centralized procedure is valid in all of the member countries of the EU. In recent years, the EMA has published several guidelines describing the conditions for authorization of both the reference product and biosimilars.51–53
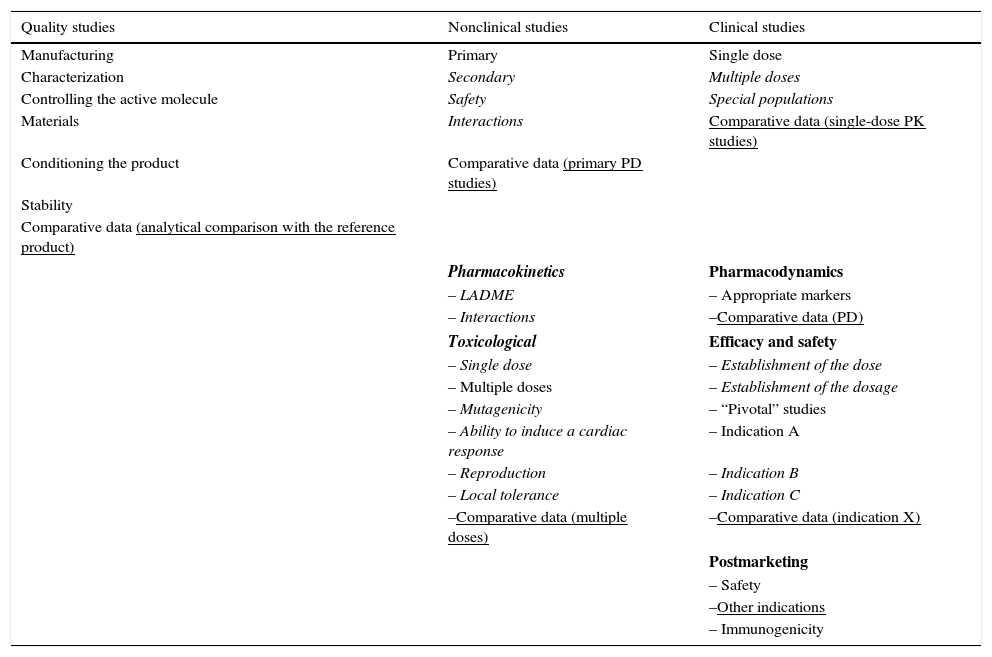
On the other hand, in the development of a biosimilar, the attempt is made to demonstrate the similarity between the biosimilar and the original drug in terms of the previously justified efficacy and safety. Thus, the procedures required for their approval are different but equally strict (Table 3). This makes it possible to reduce the financial cost and accelerate their reaching the market. Like for other medications, the pharmaceutical company must present a RMP together with the application for marketing authorization. All of the biosimilar medications in the market have a RMP with information on the plan included in the report on the evaluation published on the EMA website. The RMP of a biosimilar should consider the safety profile of the reference product. As immunogenicity is one of the subjects of interest in any biological agent, this should be dealt with in the RMP report, and may require post-authorization efficacy studies, according to the decision of the EC, for example, if there are concerns regarding certain aspects of efficacy or safety that can only be resolved once the product is commercially available.54
Requirement of the European Medicines Agency for the Approval of Innovator Biological Products and Biosimilars.
| Quality studies | Nonclinical studies | Clinical studies |
|---|---|---|
| Manufacturing | Primary | Single dose |
| Characterization | Secondary | Multiple doses |
| Controlling the active molecule | Safety | Special populations |
| Materials | Interactions | Comparative data (single-dose PK studies) |
| Conditioning the product | Comparative data (primary PD studies) | |
| Stability | ||
| Comparative data (analytical comparison with the reference product) | ||
| Pharmacokinetics | Pharmacodynamics | |
| – LADME | – Appropriate markers | |
| – Interactions | –Comparative data (PD) | |
| Toxicological | Efficacy and safety | |
| – Single dose | – Establishment of the dose | |
| – Multiple doses | – Establishment of the dosage | |
| – Mutagenicity | – “Pivotal” studies | |
| – Ability to induce a cardiac response | – Indication A | |
| – Reproduction | – Indication B | |
| – Local tolerance | – Indication C | |
| –Comparative data (multiple doses) | –Comparative data (indication X) | |
| Postmarketing | ||
| – Safety | ||
| –Other indications | ||
| – Immunogenicity | ||
Normal print: usual requirements for both products; in italics: requirements exclusively for the reference product; underlined: requirements exclusively for the biosimilars.
LADME, liberation, absorption, distribution, metabolism and elimination; PD, pharmacodynamics; PK, pharmacokinetics.
In general, biological medicines are more costly than small molecule drugs and properly managing their use is an increasingly important objective for commercial payers. Biosimilars can be a less expensive alternative to the existing biological products whose patents have expired and could favor healthy competition, thus allowing access to biological agents of a greater number of patients, contributing to the financial sustainability of health systems. Although, in absolute values, the difference in price between biosimilars and their corresponding reference drugs has less impact than that of generic drugs, the use of biosimilars has grown in recent years, clearly reducing the cost of biological therapies.55
ConclusionsPharmacovigilance systems are based on monitoring and evaluation of all the information related to a drug in order to estimate its safety once it is on the market. Traceability is an indispensable tool for this, as it enables us to reconstruct the history of a drug throughout its entire “life cycle”, making it possible to detect events, both intrinsic and extrinsic, that could affect the medication and vary the characteristics of the product as it reaches its final destination: the patient.
To report adverse drug reactions (ADR) relative to any biological agent—innovator or biosimilar—is of vital importance to clearly identify the medication involved. Thus, European legislation requires that any notification of an AE concerning a biological drug be accompanied by the approved name of the medicine and batch number, which must be guaranteed by the local authorities of each member state of the EU.56 As we can see, traceability becomes especially interesting in the field of biological drugs, as is recognized by the competent authorities.
For this report, we reviewed the major aspects related to both processes, applied to the world of biological products. Likewise, we approached concepts such as interchangeability, immunogenicity, nomenclature and the guidelines that regulate the use of these products. Finally, we consider this to be a fascinating field for clinicians. It is continuously expanding, and is the subject of new concepts and constant changes from both basic science and the drug industry, clinical practice and health agencies. There must be proper interaction among all of these entities to achieve the most suitable and cost-effective objectives.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of InterestThe authors declare they have no conflict of interest.
The authors thank all of the members of the Pharmacy Department of Hospital de La Princesa, Madrid, Spain, for their constructive comments and appraisals that helped to improve this report.Supplementary Data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.reumae.2016.05.014.
The following are the supplementary data to this article:
Please cite this article as: Serra López-Matencio JM, Morell Baladrón A, Castañeda S. Fármacos biosimilares: un nuevo escenario en las terapias biológicas. Reumatol Clin. 2017;13:287–293.


