




Sjögren's disease it's a heterogeneous and complex rheumatological disease, can present central neurological manifestations, with a prevalence that varies between 1–5% according to the international literature. We report a series of three cases; we present a patient who debuted with epileptic seizures, one with catatonic syndrome and a last one with optic neuritis. Knowing the various presentations of the central neurological manifestations allows us to broaden the diagnostic suspicion from the beginning, providing timely treatment.
La enfermedad de Sjögren (ES) es una enfermedad reumatológica heterogénea y compleja, que puede presentar manifestaciones neurológicas a nivel central, con una prevalencia que varía entre 1–5%. Reportamos una serie de tres casos, se presenta a una paciente que debutó con crisis epilépticas, una con un cuadro de síndrome catatónico y una última con un cuadro de neuritis óptica, asociados a la ES de reciente diagnóstico. Identificar las diversas presentaciones de las manifestaciones neurológicas centrales, permite ampliar la sospecha diagnóstica desde un inicio, brindando así un tratamiento oportuno.
Sjögren's disease (SS) is a chronic systemic autoimmune disease, characterised by lymphocytic infiltration, mainly affecting the lacrimal and salivary glands. The incidence is more frequent in women, with a 6:1 ratio, and a higher prevalence among 50–60 years of age.1 Pathophysiologically, abnormalities of the innate and adaptive immune system are found, resulting in polyclonal hyperactivity of B cells and production of autoantibodies such as antinuclear antibodies (ANA), anti-Ro/SS-A and anti-La/SS-B, clinically manifesting xerophthalmia, xerostomia and other conditions.
This diagnosis should be suspected in all those persons who persistently present dry eye and/or mouth, parotid enlargement or positive serological tests such as the presence of anti-Ro/SS-A antibodies with or without anti-la/SS-B antibodies, rheumatoid factor, positive ANA and hyperglobulinemia.3 ANA titration is the screening test in patients who are suspected of SS, producing a fine mottling pattern which is the appearance most often associated with this condition. In some clinical contexts, rheumatoid factor and anticentromere antibodies may also be positive and considered within the diagnostic criteria.4
In 2016, the American College of Rheumatology and the European Alliance of Rheumatology Associations (ACR-EULAR) agreed on a new set of criteria for the classification of SS. These criteria are based on the sum of 5 elements: anti-Ro/SS-A positivity; focal lymphocytic sialadenitis with a score greater than 1; each scored as 3; abnormal eye staining score ≥ 5 (or van Bijsterveld ≥ 4 score); a Schirmer test result ≤ 5 mm/5 min, and an unstimulated salivary flow rate ≤ 0.1 ml/min, each with a score of 1. These criteria are not used on a daily basis in daily practice.3 In addition, the use of magnetic resonance imaging and ultrasound have been described to detect significant abnormalities of the glandular parenchyma.4 Some of the systemic manifestations include the nervous system, both peripheral and central.
The prevalence of neurological manifestations ranges from 8 to 49%, with an average of 20%.5 As for peripheral manifestations, these are present in 2%–60% of cases, with an average of 20%. This involvement is much more common than that of the central nervous system (CNS). The most common types of SS-related peripheral neuropathies are distal axon polyneuropathies. The usual pattern of polyneuropathy is axonal, rather than demyelinating, with disproportionate sensory involvement. As symptoms progress, patients suffer from impaired balance: they may begin with gait instability and tend to progress to severe incapacitation due to ataxia and wheelchair confinement.6 On physical examination, there is an absence of vibrational sensation, deterioration in position, positive Romberg and absence of osteotendinous reflexes. Differential diagnosis must distinguish from ataxic or sensory Guillain Barré syndrome, vitamin deficiency, chemotherapy-associated neuropathy, hereditary disorders, paraneoplastic syndrome, and spinal disease.2 In 1986 Dr. Alexander described a series of 20 cases with symptoms similar to those of multiple sclerosis. Analysis has found that central neurological manifestations precede the diagnosis of SS by up to 7 years. Since then, a clinical spectrum has been described in its presentation ranging from optic neuritis, seizures and bipolar disorder to meningitis. Any structure of the CNS can be affected, from the spinal cord, optic nerves, brainstem, and cerebral hemispheres to the cerebellum. The most common clinical manifestations are aseptic meningitis, seizures, headache, cognitive impairment, transverse myelitis, optic neuritis, ataxia, encephalopathy, and lesions similar to systemic sclerosis. Cognitive manifestations mainly show deterioration of visuospatial function, executive function, attention and memory.1
Magnetic resonance imaging (MRI) does not usually produce any findings, while the use of single-photon emission tomography (SPECT) has been more popular, in which temporal and frontal hypoperfusion has been described.4 Headache, meningeal involvement, seizures, and cranial nerve palsy are the most common signs in symptomatic cases. Multiple sclerosis-like lesions present clinically with limb paresis, speech disturbances, intranuclear ophthalmoplegia and ataxia. In the cerebrospinal fluid, oligoclonal bands and an increased IgG level are identified. MRIs show hyperintense T2 lesions in the cervical region.6
The standard treatment for SS-associated neuropathy is immunosuppressive, however between 40–50% of cases may present resistance, so starting immunotherapy early before axonal degeneration occurs is insisted upon. It has been indicated that the ideal time to start treatment is within 15 days of the onset of symptoms.7 The presence of neuropathy has been established as a major factor in the poor prognosis of the disease.
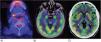
Clinical observationCase 1: A 68-year-old female patient began with loss of alertness, disorientation, delusions, visual hallucinations, and tonic-clonic seizures. The brain MRI identified lesions in the frontal lobes with punctate and hyperintense characteristics in T2 and flair sequences, as well as decreased frontal cortical brain volume. During her hospital stay, she presented gelastic crises, for which an electroencephalogram was performed which reported: abnormal stroke with diffuse dysfunction, ictal characteristics, electrical onset, recruiting rhythm in the delta range followed by tip activity in bilateral posterior regions, with right predominance, for which treatment with levetiracetam was initiated and anti-Smith, anti-SS-A and anti-SS-B negative antibodies were determined. Total positive ANA was 1:640, with a dense, fine, mottled pattern. PET-CT was performed as a supplement to the approach, in which generalised cerebral hypometabolism was reported, as well as increased basal ganglia uptake (Fig. 1A). Salivary glandular biopsy was performed with a report of atrophy of mucinous acini with a focus of moderate lymphoid infiltrate of more than 50 lymphocytes, as well as data on fibrosis, ductal ectasia and abundant apoptotic bodies (Fig. 2). Management was started with boluses of methylprednisolone and later rituximab, presenting clinical improvement.

(A) Positron emission tomography (PET-CT) of case 1. Changes in salivary gland density were observed that correlate with Sjögren's disease, with an increased metabolism focus in the right submandibular gland of 10 mm with a SUVmax of 5.8. B and C) PET-CT for case 2. Increased metabolism was observed in both hippocampi, the right temporal cortex and both putamens. The rest of the cortex was observed to have generalised hypometabolism.

(A) Biopsy of the minor salivary gland with atrophy of mucinous acini with a focus of moderate lymphoid infiltrate of more than 50 lymphocytes surrounding the dilated ducts, as well as evidence of fibrosis, ductal ectasia and abundant apoptotic bodies. Hematoxylin-eosin staining, ×10. B) Mild periductal lymphocytic infiltration of more than 50 lymphocytes per field. Hematoxylin-eosin stain ×40.
Case 2: A 20-year-old female patient with no previous comorbidities, who presented with sudden disorganised thinking, poor speech content, delirium of persecution, auditory hallucinations and disorientation, as well as catatonic syndrome, characterised by hypomimia, mutism, anergy and apathy. She presented a suicide attempt with a high degree of lethality, with subsequent pharmacological management by Psychiatry. A PET-CT scan was performed, which documented a lateral occipital cortex with decreased metabolism, caudate nuclei and putamen with metabolic increase, as well as cerebellum with generalised hypometabolism (Fig. 1B and C). Autoimmune encephalitis antibodies were negative. Total ANA with titration was 1:320 with a dense fine mottled pattern, anti-DNA DS recorded 34.5 IU/ml, anti-SS-A: 153.6 IU/ml and anti-SS-B negative. Treatment with methylprednisolone boluses was initiated. Four months after her discharge, she presented akinetic mutism, inability to perform daily activities and discoordination, for which plasmapheresis was performed, with improvement in the condition.
Case 3: A 41-year-old female patient with a pregnancy of 11.3 weeks of gestation, which began with abrupt loss of vision in the right eye in the central region, accompanied by pain when performing eye movements. Lumbar puncture was undertaken, detecting the presence of multiple oligoclonal bands. An MRI of the skull and spinal cord was performed, in which an increase in fluid in the optic nerve with right predominance was observed, as well as visual evoked potentials, where a slight dysfunction of the right visual pathway was reported, due to a decrease in the number of functional fibres and predominance of central vision with prechiasmatic location.
Anti-MOG and anti-aquaporin 4 (AQP4) antibodies showed a negative result. Due to atypical presentation, total positive ANA was requested: 1:320, presenting a fine mottled pattern, anti-SS-A positive at 790 IU/mL and anti-SS-B negative, with the rest negative. Management with rituximab and 5 plasmapheresis sessions were initiated, with clinical improvement.
DiscussionSeveral articles have discussed the presence of peripheral neurological manifestations in SS. In contrast, central neurological symptoms are identified less frequently, some of those described being: seizures, neuritis, strokes, alterations in cognitive function and encephalopathy, as was observed in this series of cases. In a cross-sectional study, Fujioka et al.2 analysed 512 patients diagnosed with SS, finding a prevalence of neurological manifestations of 46%, associated with the male sex, advanced age at onset of the disease and hospitalisation at the first presentation. Popescu et al.5 concluded that the highest prevalence of central neurological manifestations in SS are aseptic meningitis, changes in behaviour, and cognitive impairment. Fana et al.4 identified pulmonary involvement, anti-SS-A positivity and low C3 levels as prognostic factors for involvement of SS in the CNS. In this case series, different central neurological manifestations presented that were the first symptom of SS, and this can be a confounding factor that could inspire us to reach a precise diagnosis. These clinical manifestations have a significant impact on quality of life and significant diagnostic difficulty, so neurologists and rheumatologists must work together to identify them. Identifying atypical symptoms in a timely manner enables effective treatment to be provided to avoid other complications.
ConclusionsThe presence of neurological manifestations in the absence of systemic symptoms rarely leads us to consider SS as a presumptive diagnosis, so the description of this series of cases may guide health care professionals to consider it as a diagnostic suspicion and be able to offer targeted treatment that would provide patients with an improvement in prognosis and quality of life.
Ethics of scientific publishingAll personal data were protected under the confidentiality of the main author, who obtained the signature of informed consent from all of the patients involved.
FundingThis research has not received any specific grants from public sector agencies, the commercial sector or non-profit entities.
The authors have no conflict to declare.


