



La enfermedad de Sjögren (ES) es una enfermedad reumatológica heterogénea y compleja, que puede presentar manifestaciones neurológicas a nivel central, con una prevalencia que varía entre el 1-5%. Reportamos una serie de 3 casos, en la que se presenta a una paciente que comenzó con crisis epilépticas, una con un cuadro de síndrome catatónico y la última con un cuadro de neuritis óptica, asociados a la ES de reciente diagnóstico. Identificar las diversas presentaciones de las manifestaciones neurológicas centrales permite ampliar la sospecha diagnóstica desde un inicio, brindando así un tratamiento oportuno.
Sjögren's disease it's a heterogeneous and complex rheumatological disease, can present central neurological manifestations, with a prevalence that varies between 1-5% according to the international literature. We report a series of three cases; we present a patient who debuted with epileptic seizures, one with catatonic syndrome and a last one with optic neuritis. Knowing the various presentations of the central neurological manifestations allows us to broaden the diagnostic suspicion from the beginning, providing timely treatment.
La Enfermedad de Sjögren (ES) es una enfermedad autoinmune sistémica crónica, que se caracteriza por la infiltración linfocítica, afectando principalmente a las glándulas lagrimales y salivales. La incidencia es más frecuente en mujeres, con una relación 6:1, y una mayor prevalencia entre los 50-60 años1. Fisiopatológicamente se encuentran anomalías del sistema inmunológico innato y adaptativo, que resulta en una hiperactividad policlonal de las células B y producción de autoanticuerpos como anticuerpos antinucleares (ANA), anti-Ro/SS-A y anti-La/SS-B, manifestándose clínicamente xeroftalmia, xerostomía y otras2.
El diagnóstico debe sospecharse en todas aquellas personas que presenten de manera persistente ojo y/o boca secos, agrandamiento de parótidas o pruebas serológicas positivas como presencia de anticuerpos anti-Ro/SS-A con o sin anticuerpos anti-la/SS-B, factor reumatoide, ANA positivos e hiperglobulinemia3. La titulación de ANA es la prueba de cribado en los pacientes que se tenga la sospecha de ES, siendo el patrón de moteado fino el más asociado. En algunos contextos clínicos el factor reumatoide y los anticuerpos anticentrómero también pueden ser positivos y considerarse dentro de los criterios diagnósticos4.
En 2016, el Colegio Estadounidense de Reumatología y la Alianza Europea de Asociaciones de Reumatología (ACR-EULAR, por sus siglas en inglés) acordaron un nuevo conjunto de criterios para clasificación de la ES; estos se basan en la suma de 5 elementos: positividad de anti-Ro/SS-A, sialoadenitis linfocítica focal con puntuación mayor a 1, cada uno con una puntuación de 3; puntuación de tinción ocular anormal≥5 (o puntuación de van Bijsterveld≥4), un resultado de la prueba de Schirmer≤5mm/5min y una tasa de flujo salival no estimulada≤0,1ml/min, cada una con una puntuación de 1. Estos criterios no se utilizan de manera cotidiana en la práctica diaria3. De manera adicional se ha descrito el uso de la resonancia magnética y de la ecografía para detectar anomalías significativas del parénquima glandular4. Algunas de las manifestaciones sistémicas incluyen el sistema nervioso, tanto periférico como central.
La prevalencia de manifestaciones neurológicas oscila entre el 8-49%, con una media del 20%5. En cuanto a las manifestaciones periféricas se presentan del 2% al 60%, con un promedio de 20%. Esta afectación es mucho más frecuente que la del sistema nervioso central (SNC). Los tipos más comunes de neuropatías periféricas relacionadas con la ES son las polineuropatías axónicas distales. El patrón habitual de polineuropatía es axonal, en lugar de desmielinizante, con afectación sensitiva desproporcionada. A medida que los síntomas progresan los pacientes sufren un deterioro del equilibrio: pueden comenzar con inestabilidad de la marcha y tienden a progresar a una incapacitación grave por ataxia y confinamiento en silla de ruedas6. En la exploración física hay ausencia de sensación vibratoria, deterioro de la posición, Romberg positivo y ausencia de reflejos osteotendinosos. Se tiene que establecer el diagnóstico diferencial con el síndrome de Guillain Barré atáxico o sensitivo, deficiencia de vitaminas, neuropatía asociada a quimioterapia, trastornos hereditarios, síndrome paraneoplásico y enfermedad de columna2. En 1986 el Dr. Alexander describió una serie de 20 casos con síntomas similares a los de la esclerosis múltiple. Se ha analizado que las manifestaciones neurológicas centrales preceden al diagnóstico de ES hasta 7 años. Desde entonces se ha descrito un espectro clínico en su presentación que va desde neuritis óptica, convulsiones y trastorno bipolar hasta meningitis. Cualquier estructura del SNC puede verse afectada, desde la médula espinal, los nervios ópticos, el tronco encefálico y los hemisferios cerebrales hasta el cerebelo. Las manifestaciones clínicas más comunes son meningitis aséptica, convulsiones, cefalea, deterioro cognitivo, mielitis transversa, neuritis óptica, ataxia, encefalopatía y lesiones similares a la esclerosis sistémica. En cuanto a las manifestaciones cognitivas se presenta principalmente deterioro de la función visoespacial, ejecutiva, de la atención y la memoria1.
La resonancia magnética (RM) no suele tener ningún hallazgo, mientras que el uso de tomografía por emisión de fotón único (SPECT, por sus siglas en inglés) ha tenido mayor auge, en el que se ha descrito hipoperfusión temporal y frontal4. La cefalea, afectación meníngea, convulsiones y parálisis de los nervios craneales son los signos más comunes en los casos sintomáticos. En cuanto a las lesiones similares a la esclerosis múltiple se presentan clínicamente con paresia de las extremidades, alteraciones del habla, oftalmoplejía intranuclear y ataxia. En el líquido cefalorraquídeo se identifican bandas oligoclonales e índice IgG aumentado. En la RM se aprecian lesiones hiperintensas en T2 en la región cervical6.
El tratamiento estándar de la neuropatía asociada a ES es la inmunosupresora, sin embargo entre el 40-50% de los casos pueden presentar resistencia, por lo que se ha insistido en iniciar inmunoterapia de manera temprana antes de que se produzca degeneración axonal. Se ha indicado que el tiempo idóneo de comienzo del tratamiento es antes de 15 días de iniciados los síntomas7. Se ha establecido como un fuerte factor de mal pronóstico de la enfermedad la presencia de neuropatía.
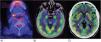
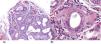
Observación clínicaCaso 1: paciente mujer de 68 años de edad, que comenzó con pérdida del estado de alerta, desorientación, ideas delirantes, alucinaciones visuales y crisis tónico-clónica. En la RM de encéfalo se identificaron lesiones en los lóbulos frontales de características puntiformes e hiperintensas en secuencias T2 y flair, así como disminución del volumen cerebral cortical de tipo frontal. Durante su estancia hospitalaria presentó crisis gelásticas, por lo que se le realizó un electroencefalograma en el que se reportó: trazo anormal con disfunción difusa, de características ictales, de inicio eléctrico, de ritmo reclutante en rango delta seguido por actividad de puntas en regiones posteriores bilaterales, con predominio derecho, por lo que se inició tratamiento con levetiracetam y se determinaron anticuerpos anti-Smith, anti-SS-A y anti-SS-B negativos, ANA totales positivos 1:640 con patrón moteado fino denso. Se realizó PET-TC como complemento del abordaje, en el que se reportó hipometabolismo cerebral generalizado, así como incremento en la captación de ganglios basales (fig. 1A). Se realizó biopsia glandular salival con reporte de atrofia de acinos mucinosos con foco de infiltrado linfoide moderado de más de 50 linfocitos, así como datos de fibrosis, ectasia ductal y abundantes cuerpos apoptóticos (fig. 2). Se inició manejo con bolos de metilprednisolona y posteriormente rituximab, presentando mejoría clínica.

A) Tomografía por emisión de positrones (PET-TC) del caso 1. Se observa alteración en la densidad de las glándulas salivales que se correlaciona con enfermedad de Sjögren, con un foco de metabolismo incrementado en la glándula submandibular derecha de 10mm con SUVmáx de 5,8. B y C) PET-TC del caso 2. Se observan, con aumento del metabolismo en ambos hipocampos, la corteza temporal derecha y ambos putámenes. El resto de la corteza se observa con hipometabolismo generalizado.

A) Biopsia de glándula salival menor con atrofia de acinos mucinosos con foco de infiltrado linfoide moderado de más de 50 linfocitos que rodean los conductos dilatados, así como datos de fibrosis, ectasia ductal y abundantes cuerpos apoptóticos. Tinción hematoxilina-eosina, ×10. B) Infiltración linfocítica periductal leve de más de 50 linfocitos por campo. Tinción hematoxilina-eosina ×40.
Caso 2: paciente mujer de 20 años de edad sin comorbilidades previas, quien presentó de manera súbita pensamiento desorganizado, contenido pobre del discurso, delirio de persecución, alucinaciones auditivas, desorientación, así como síndrome catatónico, caracterizado por hipomimia, mutismo, anergia y apatía. Presentó un intento suicida de alta letalidad, con posterior manejo farmacológico por parte de psiquiatría. Se le realizó PET-TC en la que se documentó corteza occipital lateral con disminución en el metabolismo, núcleos caudado y putámenes con incremento metabólico, así como cerebelo con hipometabolismo generalizado (figs. 1B y C). Anticuerpos de encefalitis autoinmune negativos. ANA totales con titulación 1:320 con patrón moteado fino denso, anti-DNA DS 34,5UI/ml, anti-SS-A 153,6UI/ml y anti-SS-B negativos. Se inició tratamiento con bolos de metilprednisolona. Cuatro meses después de su egreso presentó mutismo acinético, incapacidad para la realización de las actividades diarias e incoordinación, por lo que se le realizó plasmaféresis, con mejoría del cuadro.
Caso 3: paciente mujer de 41 años de edad, con embarazo de 11,3 semanas de gestación, que inició con pérdida abrupta de visión en el ojo derecho en la región central, acompañado de dolor a la realización de movimientos oculares. Punción lumbar con presencia de múltiples bandas oligoclonales. Se le realizó RM de cráneo y médula espinal, en la que se observa aumento de líquido en el nervio óptico de predominio derecho, así como potenciales evocados visuales, donde se reportó unaleve disfunción de la vía visual derecha por disminución del número de fibras funcionales y predominio de visión central con localización prequiasmática. Anticuerpos anti-MOG y anti-acuaporina 4 (AQP4) con resultado negativo. Por presentación atípica se solicitaron ANA totales positivos 1:320 patrón moteado fino, anti-SS-A positivos a 790UI/ml y anti-SS-B negativos, el resto negativos. Se inició manejo con rituximab y 5 sesiones de plasmaféresis, con mejoría clínica.
DiscusiónDiversos artículos han discutido la presencia de manifestaciones neurológicas periféricas en la ES. En contraste, los síntomas neurológicos centrales son identificados con menor frecuencia, algunos de los descritos son: crisis convulsivas, neuritis, accidentes cerebrovasculares, alteraciones en la función cognitiva y encefalopatía, así como se observó en esta serie de casos. En un estudio transversal Fujioka et al.2 analizaron a 512 pacientes diagnosticados de ES, encontrando una prevalencia de manifestaciones neurológicas del 46%, asociándose al sexo masculino, la edad avanzada al inicio de la enfermedad y la hospitalización en la primera presentación. Popescu et al.5 concluyen que la mayor prevalencia de manifestaciones neurológicas centrales en la ES son meningitis aséptica, cambios en el comportamiento y deterioro cognitivo. Fana et al4. identificaron la afectación pulmonar, la positividad de anti-SS-A y los niveles bajos de C3 como factores pronósticos de afectación de la ES al SNC. En esta serie de casos se presentan diferentes manifestaciones neurológicas centrales que fueron el primer síntoma de la ES, lo cual puede ser un factor confusor que aliente la elaboración de un diagnóstico de precisión. Dichas manifestaciones clínicas tienen una importante repercusión en la calidad de vida y cuentan con una amplia dificultad diagnóstica, por lo que neurólogos y reumatólogos deben trabajar de manera conjunta para su identificación. Identificar oportunamente aquellos síntomas atípicos permite brindar un tratamiento efectivo para evitar otras complicaciones.
ConclusionesLa presencia de manifestaciones neurológicas en ausencia de sintomatología sistémica rara vez nos orienta a considerar como diagnóstico presuntivo la ES, por lo que la descripción de esta serie de casos podrá orientar a los profesionales de la salud a considerarlo como sospecha diagnóstica y poder ofrecer un tratamiento dirigido que brinde a los pacientes una mejoría en el pronóstico y calidad de vida.
Ética de la publicación científicaTodos los datos personales se protegen bajo confidencialidad del autor principal, quien cuenta con la firma del consentimiento informado de cada uno de los pacientes involucrados.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores no tienen ningún conflicto que declarar.


