




We describe a 42-year-old woman with inactive systemic lupus erythematosus (SLE) at the time of her visit. She presented with an atypical movement disorder and partial continuous epilepsy. A brain biopsy excluded cerebral vasculitis and tumoral processes. We discuss the differential diagnosis of status epilepticus in a patient with SLE.
Describimos a una paciente de 42 años de edad con diagnóstico de lupus eritematoso sistémico (LES) inactivo al momento de la visita. Se presentó con desórdenes del movimiento atípicos y epilepsia parcial continua (EPC). Una biopsia cerebral excluye procesos tumorales y vasculitis. Discutimos diagnósticos diferenciales de estatus epiléptico en una paciente con LES.
The case is a 42-year-old female patient, diagnosed with SLE (ACR criteria 1997) 5 years ago, with joint and hematological involvement, immunological changes and low C3. Her disease was inactive for the past 3 years (SLEDAI 0) and she was being treated with hydroxychloroquine 200mg/day.
She had no family history of neurological or rheumatic disorders. She denied toxic habits, illicit drug use or a history of drug allergy. No history of surgery or trauma was present, with 3 daughters born at terms pregnancies without obstetric complications.
She came spontaneously to the clinic on April 8, 2010 for involuntary movements of the foot and the left hand that had begun the day before, which determined her hospitalization.
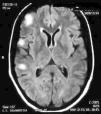
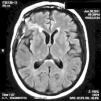
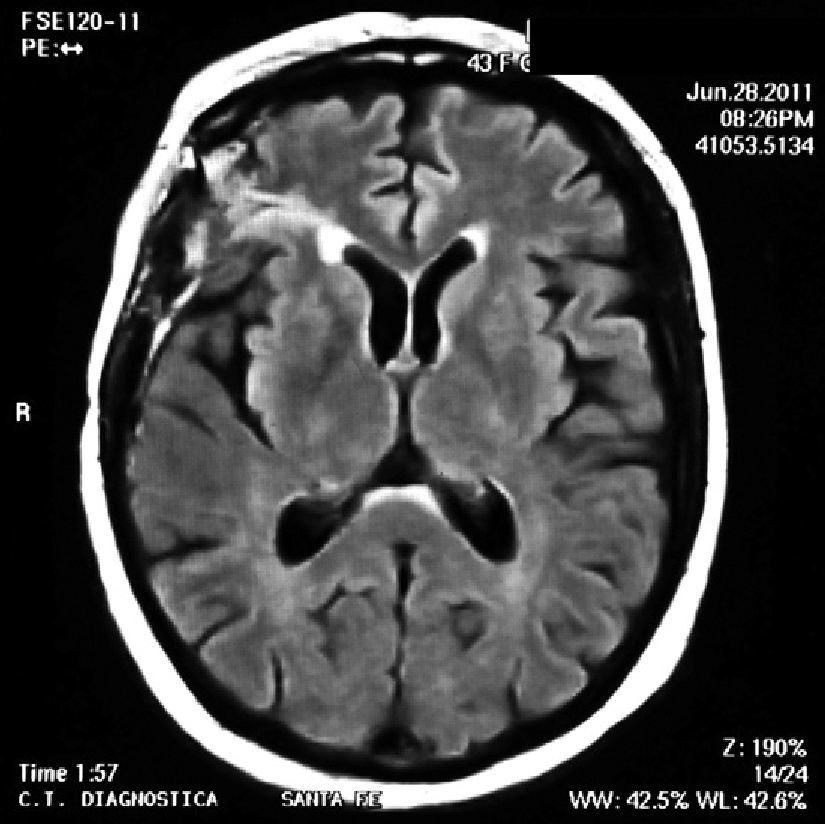
On physical examination there were no clinical signs of SLE activity or infectious diseases. Neurological examination revealed the presence of increased tendon reflexes on the left leg and a positive Babinski sign. Magnetic resonance imaging (MRI) showed T2 hyperintense lesions in the cortico-subcortical right frontal and parietal regions that stood out with gadolinium (Fig. 1). The patient began treatment with oral prednisone 50mg/day and was discharged from the hospital after 48h.
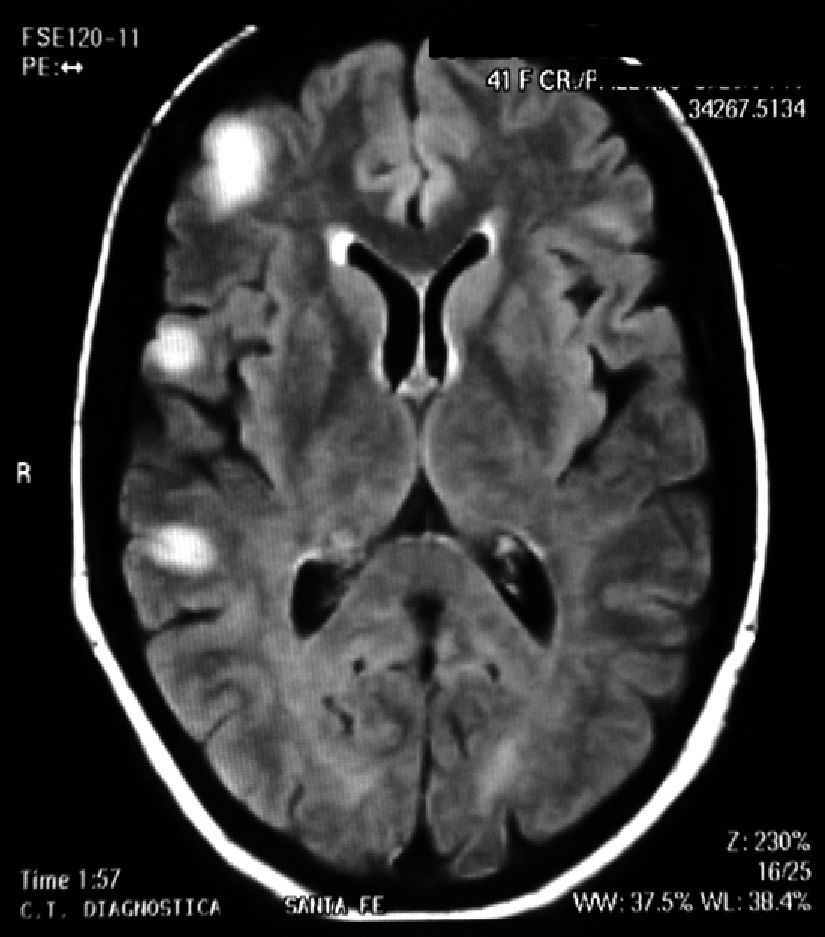
A week later (April 17), the patient reported a fall after loss of consciousness, for which she was readmitted to the hospital with the conclusion that she had a generalized seizure. On neurological examination she was found to have rhythmic movements, persistent hemiparesis of her left lower limb (foot) and positive Babinsky.
During an electroencephalogram (EEG) she developed a tonic-clonic generalized seizure with loss of consciousness and postictal coma with ad integrum restitution. The EEG showed mild impairment of cortical bioelectrical activity, excess beta rhythms and parieto-temporal isolated sharp waves. On April 24, the patient developed a third generalized seizure.
CT scans of the brain and chest were normal. The blood count, erythrocyte sedimentation rate, renal function, anticardiolipin antibodies, lupus anticoagulant, C3, C4, AAN, and anti-dsDNA antibodies were normal. The blood and urine cultures were negative. A lumbar puncture was performed, resulting in a cerebrospinal fluid (CSF) with a normal cytochemical analysis, with negative cultures (including fungi, bacteria, mycobacteria tuberculosis, Cryptococcus, Nocardia, and Listeria). We also performed polymerase chain reaction (PCR) for varicella zoster, herpes simplex, and Epstein-Barr virus, which was negative in all cases.
Seizures located on the left foot continued despite treatment with immunosuppressive intravenous methylprednisolone (3 pulses of 1g/day for 3 on consecutive days) and azathioprine 50mg/day, and the additive treatment of antiepileptic drugs such as pregabalin, carbamazepine, phenytoin, and valproic acid. There was a hypersensitivity syndrome with skin reactions, fever, and elevated transaminase levels. Suspecting a skin drug reaction, these were suspended and a skin biopsy was performed. It reported erythema multiforme with negative cultures and therefore it was assumed as an adverse drug reaction. By suspending anticonvulsant drugs, clonazepam was employed at a dose of 2g/day orally as well as IV immunoglobulin (2g/kg for 5 consecutive days and then 4 monthly doses of 0.5g/kg). The seizures stopped after a month of starting treatment.
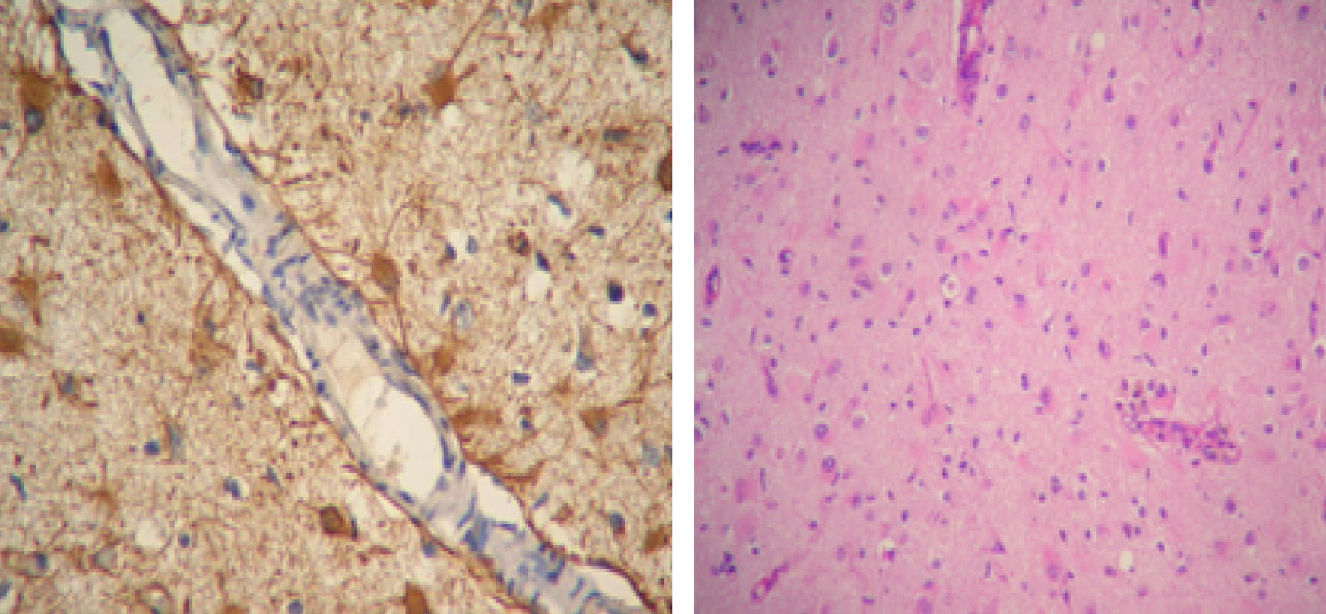
SLE remained inactive and the prednisone dose was gradually lowered. In March 2011 (after 10 months of being neurologically asymptomatic) she showed cognitive deterioration. A brain MRI revealed hyperintense lesions on T2 with the same features as above. We indicated a new pulse of 30mg IV gamma globulin without improving the clinical response and with partial epilepsy still present, this time of the right leg. We performed a new brain MRI that showed lesions which had now become bilateral. We decided to perform a biopsy of the right frontal lobe brain which reported: cortical nervous parenchyma with increased cellularity at the expense of a reactive astrogliosis. No vasculitis or neoplastic proliferation was seen. Immunostaining: GFAP+CD68+NOGO A (−), P53 (−), IDH1 (−) (Fig. 2).
Treatment was initiated with 1g of IV cyclophosphamide, which resulted in restitution ad integrum of the lesions (Fig. 3) with clinical improvement after the first pulse.
At present, the patient has inactive SLE and is free of seizures while being treated with prednisone, hydroxychloroquine 200mg/day, clonazepam 1mg/day, and monthly pulse IV cyclophosphamide.
Differential Diagnosis (Dr. Sergio Paira)When faced with a patient with SLE presenting seizures, the first step in diagnostic reasoning should be to ask whether there is a secondary underlying pathology and concomitant conditions the treatment thereof.
Seizures in Lupus ErythematosusSeizures are considered as an important manifestation of neuropsychiatric SLE.1 Between 10% and 20% of SLE patients develop epileptic seizures at some point in their illness. This means there is almost an 8 times higher prevalence of epilepsy than in the general population, therefore, it is much more common in SLE patients than expected. Between 5% and 10% of patients have mild seizures that can occur even years before the clinical onset of SLE.2
Seizures have been reported in up to 24%–45% of patients with SLE with neurological involvement, but in the Hopkins lupus cohort only 10% were affected. Generally, seizures are produced in the context of an active systemic disease, but may be an isolated finding. Other possible causes of seizures are infections, uremia, hypertension or a previous stroke, so these conditions should always be excluded.3
About a quarter of seizures in patients with SLE are clinically detectable, with symptoms ranging from disturbances in sight to focal motor deficits or sensory focal seizures, and cannot be associated with focal brain1 pathophysiological processes.
The most direct cause of epilepsy in patients with SLE is the one associated with stroke. The most common seizure types in this group of patients are usually simple partial or complex seizures with or without secondary generalization. The main causes of focal brain lesions or thrombosis are cardiac embolism, both of large vessels and the microvasculature, or primary vasculitis but these are infrequent.1
Liou et al. found that epilepsy was 3.7 times higher among patients with SLE who had anticardiolipin antibodies than among patients with SLE who did not have the antibodies.4,5
Antiphospholipid Antibodies and EpilepsyShoenfeld et al. concluded that epilepsy is common in APS and most of the risk appears to be related to extensive vascular disease manifested as central nervous system (CNS) disease, heart valve disease, livedo reticularis, and the presence of SLE.5
The clinical, EEG or focused damage images in association with ANA, suggest that vascular occlusion of small cerebral vessels may be responsible for the occurrence of seizures in some patients with SLE.
Inzelberg and Korczyn proposed that seizures in patients with APS are the expression of ischemic events that occur as a result of the hipercoagulability.6 In fact, seizures are a known symptom of cerebral ischemia and coexist with other vascular diseases in a high percentage (42.9%).7
MELAS syndrome (mitochondrial encephalomyopathy with lactic acidosis and stroke-like syndrome)
First described by Pavlakis et al. in 1984, mitochondrial encephalopathy with lactic acidosis and stroke-like syndrome (MELAS) is the convergence of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. This is a genetic disease caused by mutations of the maternal mitochondrial genome, affecting the synthesis of adenosine triphosphate (ATP). The mutations are heteroplasmic in different tissues, which could involve especially those which are highly energy-dependent, such as muscles, brain, and CNS tissues. Neurologic manifestations include speech and visual disturbances, seizures and status epilepticus. MELAS may occur with continuous partial epilepsy. Seizures are often preceded by lengthy episodes of migraine. Partial focal seizures and stroke are, in many cases, originated in the occipital lobe. This condition should always be evaluated in patients under 40 years of age with stroke, regardless of whether they have a family history of disease.8
Our patient was older than 40 years, had no family history and no lactic acidosis.
Tropical Causes of EpilepsyThere are several tropical parasitic diseases causing epilepsy.
Neurocysticercosis is the main cause of focal epilepsy starting in adulthood in endemic areas (30%–50%). All phases of cysticerci (viable, transitional, and calcified) are associated with seizures. Specific treatment promotes faster disappearance of the cysticerci and reduces the risk of seizure recurrence in patients with viable cysts.
Symptomatic epilepsy may be the first manifestation of neuroschistosomiasis in patients without systemic symptoms. The pseudotumoral form can lead to crises secondary to the presence of granulomas and edema of the cerebral cortex. Schistosoma japonicum eggs are smaller, and more easily reach the CNS and cause more frequent seizures.
Both toxocariasis and sparganosis are helminthiasis that can cause symptomatic seizures. Similarly, cerebral malaria, particularly Plasmodium falciparum, can cause chronic epilepsy.
About 20% of patients with cerebral infarction secondary to Chagas disease presented late vascular epilepsy as a complication. In this case we ruled out the presence of such pathogens.9
VasculitisCNS vasculitis represents a heterogeneous group of inflammatory diseases that primarily affect small blood vessels and leptomeningeal cerebral10 parenchyma. A variety of neurological insults can cause CNS vasculitis, including infection, cancer, ionizing radiation, cocaine use, and autoimmune10 diseases.11 Among these, the primary CNS vasculitis, SLE, polyarteritis nodosa, giant10 cell arteritis, and Sjögren's syndrome are the main autoimmune diseases associated with CNS vasculitis.12
While these disorders may present with generalized or focal seizures, our patients did not have signs of systemic vasculitis or morphological or histological evidence of cerebral vasculitis.
NeoplasmsBrain tumors, whether benign or malignant, are a dreaded cause of seizures, but are rare. These may be primary, to be denominated according to the cell lineage (astrocytoma, oligodendroglioma, glioblastoma, etc.), or metastatic tumors from primary neoplasms located in some other place. Similarly, it may be malignant or benign by differentiation. Regardless of the oncological characteristics, they can cause seizures due to cerebral irritation or mass effect may present with seizures and partial localized continual epilepsy (EPC).
While the tumors are causes of CLD in between 5% and 19% of cases, their presence was ruled out by morphological studies (MRI and brain biopsy).13
Rasmussen's EncephalitisRegarding the type of persistent movement disorder of the foot, we can define it as a focal status epilepticus, which is called EPC.
EPC, originally described by Koshewnikow is a rare form of focal epilepsy, characterized by a localized somato-motor status epilepticus, i.e., regular or irregular muscle contractions and clonic seizures affecting a limited part of the body produced for a minimum of one hour intervals of no more than 10s due to various injuries in a sector of the motor cortex.14
Rasmussen encephalitis (RE), or chronic encephalitis and epilepsy, first described in 1958, is a rare clinical syndrome characterized by severe focal epilepsy (continuous partial epilepsy), usually accompanied by progressive hemiparesis and cognitive impairment, that develops in association with pathological features of chronic encephalitis.15
Except for a few case reports, the frequency of association between Rasmussen's syndrome and autoimmune diseases (Sjögren's syndrome and SLE) remains uncertain.16,17
Clinical Diagnosis (Dr. Sergio Paira)Our patient had clinical features of a continuous RE with intractable partial epilepsy and hemiparesis. Radiologically we found cortical–subcortical hyperintense lesions on T2, not highlighted with gadolinium, and histological evidence of brain swelling. Therefore, having ruled out other causes of CLD, I assume that the patient has an autoimmune disease associated to RE as the primary disease.
Pathology Discussion (Dr. Susana Roverano–Dr. Gustavo Saredo)A brain biopsy is not necessary in all cases of RE, because other criteria may be sufficient to diagnose the disease, and this is reserved for cases of doubtful clinical diagnosis or atypical presentations of RE. It should be taken from an area where the T2/FLAIR signal is increased in the MR.18 In cases in which MRI lesions are not clear, other studies such as PET or SPECT may be useful in determining the site of the biopsy.19
A German group described inflammatory cells and reactive astrocytes in brain samples obtained from regions with abnormalities on MRI. In areas where hyperintensity was observed, increased numbers of T cells, nodules of microglia and GFAP+ astrocytes are regarded as the chronically most affected areas.18
Another relevant diagnostic observation is that less than 5% of CD68+ cells had macrophage morphology; the vast majority had microglial20 morphology.
These histopathological changes are similar to those observed in our patient, so the final diagnosis was that of RE in a patient with SLE.
Final Results and CommentsAlthough RE has long been considered a childhood disease (it has a more rapid and severe onset), adolescent and adult patients (with a more protracted and milder course, with a long and relatively nonspecific prodromal phase) have been described by several groups on the basis of the study of Hart et al., estimated at about 10% of cases of RE.21–24
Clinically, RE is characterized by intractable focal seizures, called EPC, and cognitive impairment secondary to hemispheric involvement.22
There have been three special features of epilepsy in patients with RE: (a) the polymorphism of seizures in a given patient, (b) refractory to medical treatment of seizures, especially EPC, and (c) simple partial motor seizures involving one side of the body (77% of cases).
One can also see secondarily generalized tonic-clonic seizures (42%), complex partial seizures (19% with generalized and 31% with subsequent unilateral motor), postural seizures probably originating in the supplementary motor area (24%) and somatosensory seizures (21%).22,24
RE is an example of an autoimmune disease of the CNS. Serum samples from patients with this disease contain antibodies against glutamate receptors GluR3 and GluR (against the epsilon subunit 2). These patients may also have stimulated GluR epsilon 2T cells in peripheral blood, and it has also been shown that both cellular and humoral autoimmunity against GluR epsilon 2 may contribute to pathophysiological processes in RE. Their presence in other autoimmune disorders suggests similar pathological mechanisms, but not RE markers. The serum of some patients with RE also contain high levels of ‘classic’ autoimmune antibodies (glutamic acid decarboxylase, anti-cardiolipin, B2-glycoprotein I and SS-A, and RNP nuclear antigens).25
The diagnosis of Rasmussen's report is based on clinical studies, electrophysiological (EEG) and morphological (MRI and, in some cases, histopathology) .26
Our patient had two unusual features of RE, she was 42 years of age and had bilateral involvement.
Bilateral RE is very rare. Some of the clinical and electrophysiological characteristics in typical unihemispheric cases or bilateral cerebral involvement have been suggested to be, for example, as a secondary spread of focal seizures in the contralateral side, intercritical epileptiform abnormalities on the contralateral side or contralateral mild atrophy.27 Therefore, the term ‘bilateral RE’ should be reserved for cases with inflammatory lesions in both hemispheres. Until 2005, about 200 cases were reported in the literature, with only 924 having bihemispheric involvement.
There is now a wide range of available therapeutic strategies, such as bolus intravenous methylprednisolone, immunosuppressants, intravenous immunoglobulin (IVIG), plasmapheresis, cyclophosphamide, and rituximab. Surgical treatment is reserved for seizure control in refractory children and adults.24,28
Ethical disclosuresProtection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of Data. The authors declare that no patient data appears in this article.
Right to privacy and informed consent. The authors have obtained the informed consent of the patients and /or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of InterestThe authors have no disclosures to make.
Please cite this article as: Benavente E, et al. Encefalitis con estatus convulsivo localizado en una paciente con lupus eritematoso sistémico. Reumatol Clin. 2012. http://dx.doi.org/10.1016/j.reuma.2012.05.007.


