




Describimos a una paciente de 42 años de edad con diagnóstico de lupus eritematoso sistémico (LES) inactivo al momento de la visita. Se presentó con desórdenes del movimiento atípicos y epilepsia parcial continua (EPC). Una biopsia cerebral excluye procesos tumorales y vasculitis. Discutimos diagnósticos diferenciales de estatus epiléptico en una paciente con LES.
We describe a 42-year old woman with inactive systemic lupus erythematosus (SLE) at the time of her visit. She presented with an atypical movement disorder and partial continuous epilepsy. A brain biopsy excluded cerebral vasculitis and tumoral processes. We discuss the differential diagnosis of status epilepticus in a patient with systemic lupus erythematosus.
Paciente de sexo femenino, de 42 años de edad, con diagnóstico de LES (criterios ACR 1997), de 5 años de evolución, con compromiso articular, hematológico, inmunológico FAN y C3 bajo. Dicha enfermedad se encontraba inactiva los últimos 3 años (SLEDAI 0) y estaba siendo tratada con hidroxicloroquina 200mg/día.
No presentaba antecedentes familiares de trastornos neurológicos o reumáticos. Negaba hábitos tóxicos, uso de drogas ilegales o historia de alergia medicamentosa. Sin antecedentes de intervenciones quirúrgicas o traumatismos, con 3 hijas de embarazos a términos sin complicaciones obstétricas.
Concurre espontáneamente a la consulta el 8 de abril del 2010 por presentar movimientos involuntarios en el pie y la mano izquierda que se habían iniciado el día anterior, por lo cual se decide su internación en el hospital.
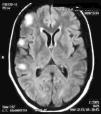
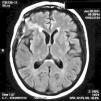
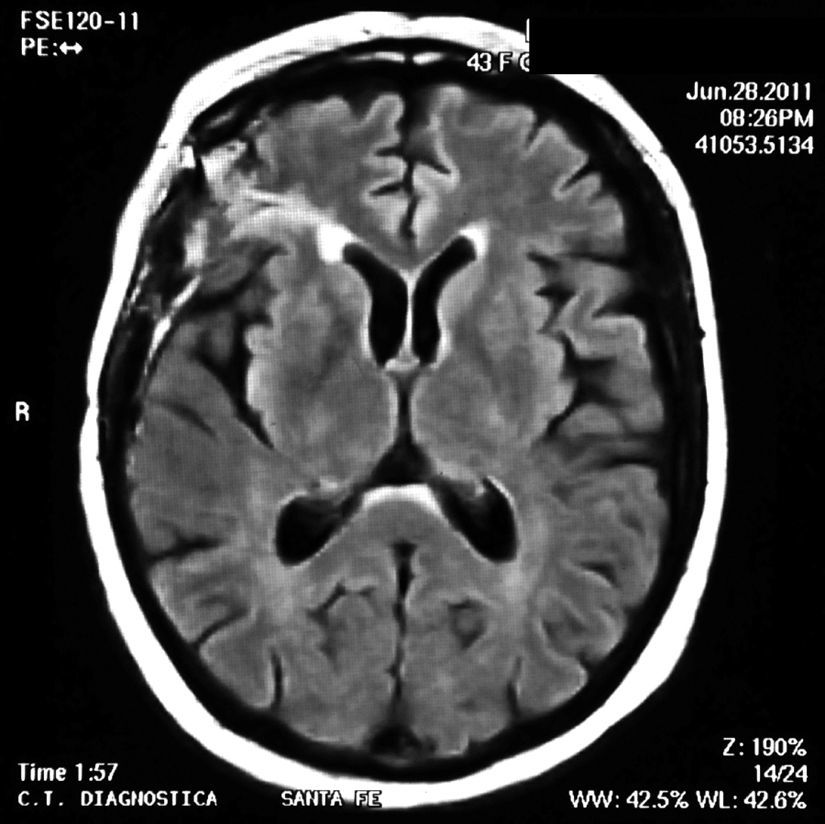
En el examen físico no había signos clínicos de actividad de LES ni de enfermedades infecciosas. El examen neurológico reveló la presencia de reflejos tendinosos aumentados en el miembro inferior izquierdo y signo de Babinsky positivo. La resonancia magnética (RM) cerebral mostró lesiones hiperintensas en T2 cortico-subcorticales en región frontal y parietal derechas que no resaltaban con gadolinio. (fig. 1). La paciente comenzó tratamiento con meprednisona 50mg/día por vía oral y fue dada de alta del hospital luego de 48 h.
Una semana después (17 de abril), la paciente informó una caída con pérdida de la consciencia, por la que fue readmitida en el hospital con la conclusión de que había tenido una convulsión generalizada. En el examen neurológico se halló hemiparesia con movimientos rítmicos persistentes en su extremidad inferior izquierda (pie) y Babinsky positivo.
Durante la realización de un electroencefalograma (EEG) desarrolló una crisis generalizada tónico-clónica con pérdida de consciencia y coma postictal con restitución ad integrum. El EEG mostró una leve alteración de la actividad bioeléctrica cortical, por exceso de ritmos beta y ondas agudas aisladas: parieto-temporales derechas. El 24 de abril la paciente presentó una tercera convulsión generalizada.
Las TC de cerebro y de tórax fueron normales. El hemograma, la eritrosedimentación, función renal, los anticuerpos anticardiolipinas, anticoagulante lúpico, C3, C4, FAN y ADNds fueron normales. Los cultivos de sangre y orina fueron negativos. Se realizó una punción lumbar, obteniéndose un líquido cefalorraquídeo (LCR) cuyo análisis citoquímico fue normal, con cultivos (hongos, gérmenes comunes, tuberculosis micobacterias, Criptococcus, Nocardia y Listeria) negativos. También se realizó reacción cadena de la polimerasa (PCR) contra la varicela zóster, herpes simple y virus de Epstein-Barr, que fue negativa en todos los casos.
Las convulsiones localizadas en el pie izquierdo continuaron a pesar del tratamiento inmunosupresor con metilprednisolona por intravenosa (3 pulsos de 1 g/día durante 3 días consecutivos) y azatioprina 50mg/día, y al tratamiento aditivo de fármacos antiepilépticos, pregabalina, carbamacepina, fenitoína y ácido valproico. Se observó un síndrome de hipersensibilidad con reacciones cutáneas, fiebre y aumento de los niveles de transaminasas. Ante la sospecha de una farmacodermia, estos fármacos fueron suspendidos y se realizó biopsia de piel. Esta informó eritema multiforme con cultivos negativos, por lo tanto, se asumió como una reacción adversa medicamentosa. Al suspender los fármacos anticomiciales se indica clonazepam 2 g/día por vía oral e inmunoglobulina IV (2 g/kg durante 5 días consecutivos y luego 4 dosis mensual de 0,5 g/kg). Las convulsiones cesaron luego de un mes del inicio del tratamiento.
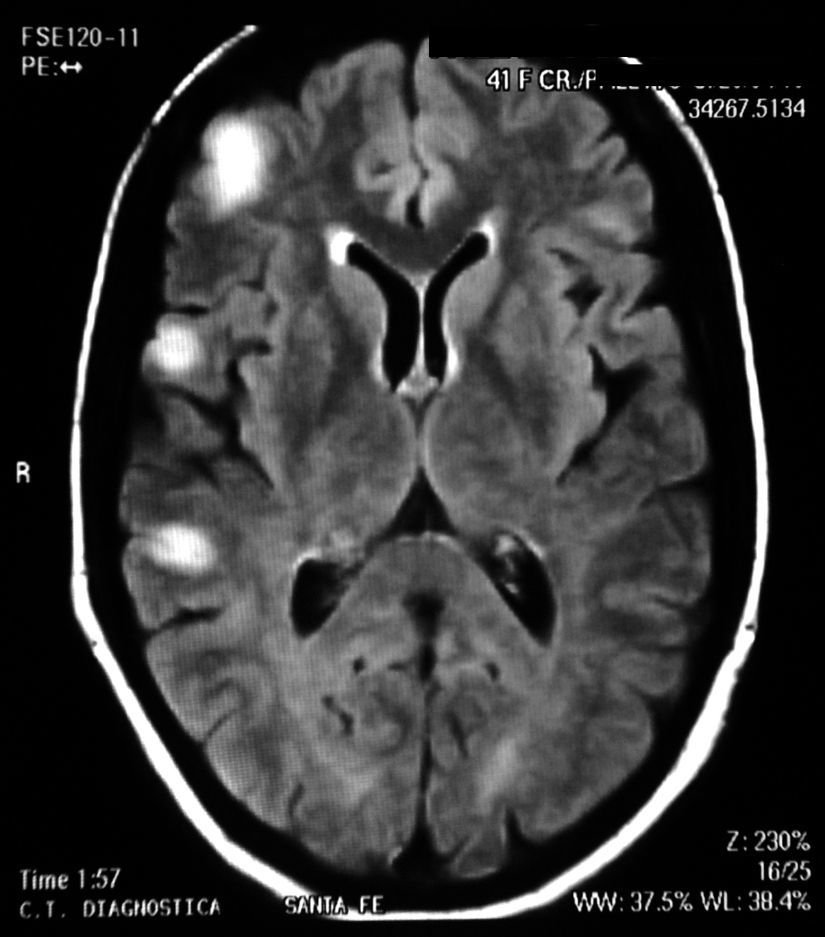
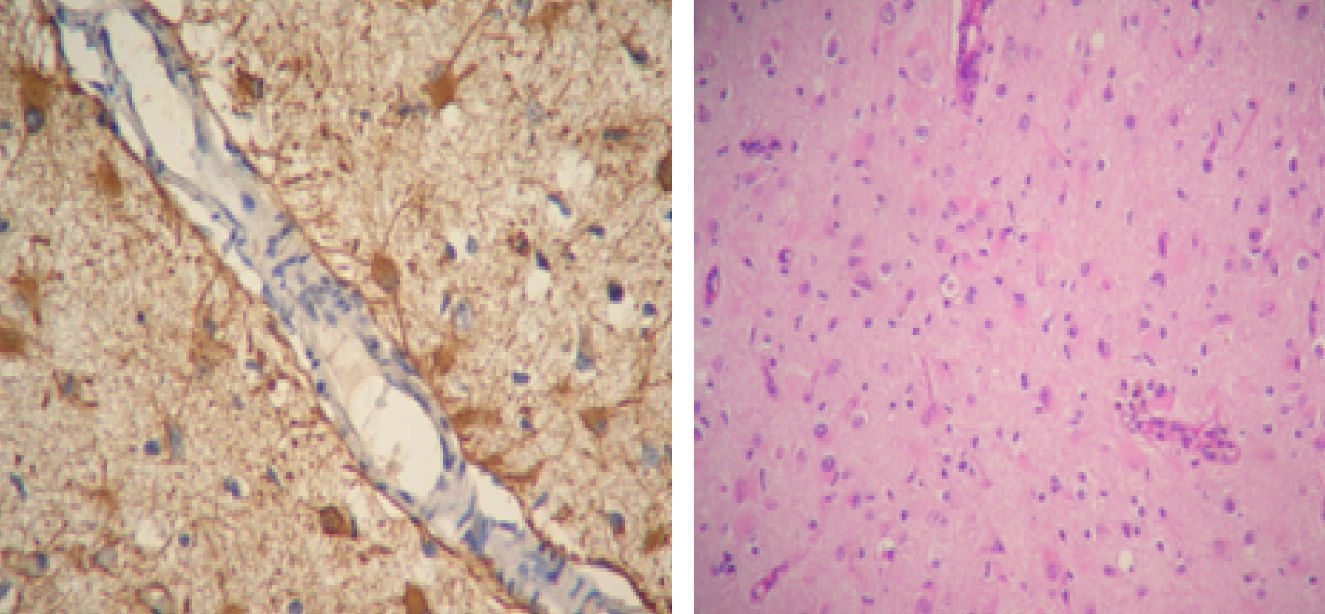
El LES permaneció inactivo y la dosis de prednisona fue descendida gradualmente. En marzo del 2011 (luego de 10 meses de encontrarse neurológicamente asintomática) presenta deterioro cognitivo. Una RM de cerebro de control revela imágenes hiperintensas en T2 de las mismas características que las anteriores. Se indica un nuevo pulso de 30mg IV de gammaglobulina, sin respuesta clínica agregándose epilepsia parcial continua, en esta oportunidad en la pierna derecha. Se realiza una nueva RM de cerebro donde se agregan nuevas lesiones las cuales se vuelven bilaterales. Se decide realizar una biopsia de cerebro del lóbulo frontal derecho que informa: parénquima nervioso cortical, con incremento de la celularidad a expensas de una astrogliosis de aspecto reactivo. No se detecta vasculitis ni proliferación neoplásica. Inmunomarcación: GFAP+ CD68+NOGO A (–), P53 (–), IDH1 (–) (fig. 2).
Se inició tratamiento con 1 g de ciclofosfamida IV que resultó en restitución ad integrum de las lesiones (fig. 3) con mejoría clínica luego del primer pulso.
En la actualidad, la paciente presenta LES inactivo y está libre de convulsiones mientras recibe tratamiento con prednisona, hidroxicloroquina 200mg/día, clonazepam 1mg/día y pulsos mensuales de ciclofosfamida.
Diagnósticos diferenciales (Dr. Sergio Paira)Ante una paciente con diagnóstico de LES que presenta convulsiones, el primer paso en el razonamiento diagnóstico que debería plantearse es si estas son secundarias su patología de base, a situaciones concomitantes o al tratamiento de la misma.
Convulsiones en el lupus eritematoso sistémicoLas convulsiones son consideradas una importante manifestación neuropsiquiátricas del LES1. Entre el 10 y 20% de los pacientes con LES desarrollan convulsiones epilépticas en algún momento de su enfermedad. Esto significa que es casi 8 veces mayor la prevalencia de epilepsia que en la población general, por lo tanto, es mucho más común en pacientes con LES de lo que cabría esperar. Entre un 5y un 10% pueden presentarse con convulsiones leves incluso años antes del inicio clínico del LES2.
Las convulsiones se han reportado hasta en un 24 a 45% de los pacientes con LES con compromiso neurológico, pero en la cohorte de lupus Hopkins solo el 10% se ven afectados. Por lo general, las convulsiones se producen en el contexto de una enfermedad sistémica activa, aunque pueden ser un hallazgo aislado. Otras posibles causas de convulsiones son procesos infecciosos, uremia, hipertensión o infartos cerebrales pasados, por lo cual dichas condiciones siempre deben ser excluidas3.
Alrededor de una cuarta parte de las convulsiones en los pacientes con LES aparecen clínicamente focalizadas al inicio, con síntomas que van desde afectaciones de la mirada con automatismos hasta déficit motor focal o convulsiones focales sensoriales, pudiendo no estar asociados a procesos fisiopatológicos focales del cerebro1.
La causa más directa de epilepsia en pacientes con LES es la que se asocia a infarto cerebral. Los tipos de crisis más frecuentes en este grupo de pacientes son, por lo general, las crisis parciales simples o complejas con o sin generalización secundaria. Las principales causas de lesiones cerebrales focales son el cardioembolismo o trombosis tanto de grandes vasos como de la microvasculatura, o la vasculitis primarias aunque estas son infrecuentes1.
Liou et al. encontraron que la epilepsia fue de 3,7 veces más frecuente entre los pacientes con LES que tenían anticuerpos anticardiolipina que entre los pacientes con LES que no tenían los anticuerpos4,5.
Anticuerpos antifosfolípidos y epilepsiaShoenfeld et al. concluyeron que la epilepsia es común en el SAF y la mayor parte del riesgo parece estar relacionado con la enfermedad vascular que se manifiesta por la afectación del sistema nervioso central (SNC) extensa, valvulopatía cardíaca, livedo reticularis, y con la presencia de LES5.
La evidencia clínica, EEG o imágenes de los daños focalizados en asociación con SAF, sugiere que la oclusión vascular de los pequeños vasos cerebrales pueden ser responsables de la aparición de convulsiones en algunos pacientes con LES.
Inzelberg y Korczyn propusieron que las convulsiones en pacientes con SAF son la expresión de los eventos isquémicos que se producen como consecuencia de la hipercoagulabilidad6. De hecho, las convulsiones son un síntoma conocido de la isquemia cerebral y la coexistencia de otras enfermedades vasculares en un alto porcentaje (42,9%)7.
Síndrome MELAS (síndrome de encefalopatía con acidosis láctica y episodios símil ictus)Descrita por primera vez por Pavlakis et al. en 1984, el MELAS (mitochondrial encephalopathy with lactic acidosis and stroke-like syndrome) es la convergencia de miopatía mitocondrial, encefalopatía, acidosis láctica y episodios tipo ictus. Esta es una enfermedad genética causada por mutaciones en el genoma mitocondrial materno, que afectan a la síntesis adenosina trifosfato (ATP). Las mutaciones tienen cargas heteroplásmica en diferentes tejidos, lo que podría implicar especialmente a los altamente dependientes de energía, tales como los músculos, el cerebro y los tejidos del SNC. Las manifestaciones neurológicas incluyen alteraciones del lenguaje y trastornos visuales, convulsiones y estado de mal epiléptico. El MELAS puede ocurrir con epilepsia parcial continua. Crisis epilépticas son precedidas frecuentemente por cuadros prolongados de migraña. Las crisis focales parciales y los accidentes cerebrovasculares son, en muchos casos, originados en el lóbulo occipital. Esta patología siempre debe ser evaluada en pacientes menores de 40 años de edad, con accidentes cerebrovasculares, sin importar si tienen antecedentes familiares con la enfermedad8.
Nuestra paciente era mayor de 40 años, sin antecedentes familiares y no presentaba acidosis láctica.
Causas tropicales de epilepsiaExisten diversas parasitosis tropicales causantes de epilepsia.
La neurocisticercosis es la principal causa de epilepsia focal de inicio en la vida adulta en áreas endémicas (30-50%). Todas las fases de los cisticercos (viables, transicionales y calcificados) se asocian a crisis epilépticas. El tratamiento específico favorece la desaparición más rápida de los cisticercos y reduce el riesgo de recurrencia de crisis en pacientes con quistes viables.
La epilepsia sintomática puede ser la primera manifestación de la neuroesquistosomiasis en pacientes sin síntomas sistémicos. La forma seudotumoral puede provocar crisis secundarias a la presencia de granulomas y edema en la corteza cerebral. Los huevos de Schistosoma japonicum son más pequeños, alcanzan más fácilmente el SNC y provocan crisis epilépticas más frecuentemente.
Tanto la toxocariasis y esparganosis son helmintiasis que pueden provocar crisis epilépticas sintomáticas. De igual forma, el paludismo cerebral, especialmente por Plasmodium falciparum puede provocar epilepsia crónica.
Alrededor del 20% de los pacientes con infarto cerebral secundario a enfermedad de Chagas presenta como complicación una epilepsia vascular tardía. En el presente caso se descartó la presencia de dichos patógenos9.
VasculitisLas vasculitis del SNC representan un grupo heterogéneo de enfermedades inflamatorias que afectan principalmente a los pequeños vasos sanguíneos leptomeníngeos y del parénquima cerebral10. Una variedad de noxas neurológicas pueden causar vasculitis del SNC, incluyendo infección, cáncer, radiación ionizante, ingesta de cocaína y enfermedades autoinmunitrias10,11. Entre estas, la vasculitis primaria del SNC, el LES, la poliarteritis nudosa, la arteritis de células gigantes10 y el síndrome de Sjögren constituyen la mayoría de las enfermedades autoinmunitarias asociadas a vasculitis del SNC12.
Si bien estos trastornos pueden presentarse con convulsiones generalizadas o focales, en nuestra paciente no se encontraron signos de vasculitis sistémica ni evidencia morfológica o histológica de vasculitis cerebral.
NeoplasiasLos tumores cerebrales, ya sean benignos o malignos, son una causa temida, de convulsiones; sin embargo, son poco frecuentes. Estos pueden ser primarios, que se van a denominar según su estirpe celular (astrocitomas, oligodendroglioma, glioblastoma etc.), o metastásicos, de algún tumor localizado en otro sitio. De igual forma, pueden ser malignos o benignos según su diferenciación. Independientemente de las características oncológicas, pueden causar crisis convulsivas por irritación cerebral o efecto de masa pudiendo presentarse con convulsiones localizadas y epilepsia parcial continua (EPC).
Si bien los tumores son causas de EPC entre un 5 y 19%, su presencia fue descartada por estudios morfológicos (RM y biopsia cerebral)13.
Encefalitis de RasmussenCon respecto al tipo de trastorno del movimiento persistente en el pie, podemos definirlo como un estatus epiléptico focalizado, lo que se da en llamar EPC.
La EPC, descripta originalmente por Koshewnikow, es una rara forma de epilepsia focal, caracterizada por un estatus epiléptico somato-motor localizado, es decir, contracciones musculares clónicas regulares o irregulares que afectan a una parte limitada del cuerpo produciéndose por un mínimo de una hora con intervalos de no más de 10 s debido a diversas lesiones de un sector de la corteza motora14.
La encefalitis de Rasmussen (ER), o encefalitis crónica y epilepsia, descrita por primera vez en 1958, es un síndrome clínico poco común, caracterizado por epilepsia focal severa (epilepsia parcial continua), por lo general acompañado de hemiparesia progresiva y deterioro cognitivo, que se desarrolla en asociación con características patológicas de encefalitis crónica15.
A excepción de algunos reporte de casos, la frecuencia de asociación entre el síndrome de Rasmussen y las enfermedades autoinmunitarias (síndrome de Sjögren y LES) sigue siendo incierto16,17.
Diagnóstico clínico del discusor (Dr. Sergio Paira)Nuestra paciente tenía las características clínicas de una ER con epilepsia parcial continua intratable y hemiparesia. Radiológicamente, lesiones córtico-subcortical hiperintensas en T2, que no resaltan con gadolinio, y evidencia histológica de inflamación cerebral. Por lo tanto, habiendo sido descartadas otras causas de EPC, asumo que la paciente presenta una ER como patología autoinmunitaria asociada a su enfermedad de base.
Discusión anatomopatológica (Dra. Susana Roverano-Dr. Gustavo Saredo)La biopsia del cerebro no es necesaria en todos los casos ER, porque otros criterios pueden ser suficientes para diagnosticar la enfermedad; esta es reservada para casos de diagnóstico clínico dudoso o presentaciones atípicas de ER. Esta debe ser tomada de un área donde se incrementa la señal T2/FLAIR en la RM18. En casos en los cuales las lesiones de la RM no son claras, otros estudios, como la PET o la SPECT, pueden ser útiles para determinar el sitio de la biopsia19.
Un grupo alemán describió células inflamatorias y astrocitos reactivos en muestras de cerebro obtenidas de las regiones con anormalidades en la RM. En las áreas donde se encontraba hiperintensidad se observó incrementado el número de células T, los nódulos de la microglía y astrocitos GFAP+con respecto a las áreas más afectadas crónicamente18.
Otra observación diagnóstica relevante es que menos del 5% de las células CD68+tenía morfología de macrófagos; la gran mayoría presentaba morfología microglial20.
Dichos cambios histopatológicos son similares a los observados en nuestra paciente, por lo cual el diagnóstico definitivo fue el de ER en una paciente con LES.
Resultado definitivo y comentarioA pesar de que la ER desde hace mucho tiempo se ha considerado una enfermedad infantil (tiene una enfermedad más rápida y severa), los pacientes adolescentes y adultos (un curso más prolongado y leve, con una larga y relativamente inespecífica fase prodrómica) han sido descritos por varios grupos y, sobre la base de las cifras de Hart et al., podría estimarse aproximadamente el 10% de los casos de ER21-24.
Clínicamente, la ER se caracteriza por convulsiones focales intratables, denominadas EPC, y deterioro cognitivo secundario a la afectación hemisférica22.
Se han observado 3 características especiales de la epilepsia en pacientes con ER: a) el polimorfismo de las convulsiones en un paciente determinado; b) la refractariedad al tratamiento médico de las convulsiones, sobre todo de EPC, y c) la crisis motoras parciales simples que implican un lado del cuerpo (77% de los casos).
Puede presentar además convulsiones tónico-clónicas generalizadas secundariamente (42%), convulsiones parciales complejas (19% con automatismo y el 31% con posterior motor unilateral), convulsiones posturales, probablemente originarias de la región motora suplementaria (24%) y convulsiones somatosensoriales (21%)22,24.
La ER es ejemplo de una enfermedad autoinmunitaria del SNC. Las muestras de suero de pacientes con esta enfermedad contienen anticuerpos contra los receptores de glutamato GluR3 y en contra de la subunidad GluR épsilon 2. Estos pacientes también pueden tener células T estimuladas por GluR épsilon 2 en circulación en sangre periférica; esto demuestra que tanto la autoinmunidad celular como la humoral contra GluR épsilon 2 pueden contribuir a los procesos fisiopatológicos en la ER. Su presencia en otros trastornos autoinmunitarios sugiere mecanismos patológicos similares, pero no son marcadores de ER. El suero de algunos pacientes con ER contiene también niveles elevados de anticuerpos autoinmunitarios «clásico» (ácido-glutámico-decarboxilasa, anti-cardiolipinas, B2-glucoproteína I y los antígenos nucleares SS-A y RNP)25.
El diagnóstico de Rasmussen se basa en estudios clínicos, electrofisiológicos (EEG) y morfológicos (RM y, en algunos casos, histopatología)26.
Nuestra paciente presenta 2 características poco comunes de ER, considerando que tiene 42 años de edad y presenta compromiso bilateral.
La ER bilateral es muy rara. Algunas de las características clínicas y electrofisiológicas en los casos típicos unihemisféricos han sugerido afectación cerebral bilateral, por ejemplo, propagación secundaria de las convulsiones focales en el lado contralateral, anormalidades epileptiformes intercríticas en el lado contralateral o atrofia leve contralateral27. Por lo tanto, el término «ER bilateral» debe reservarse para los casos con lesiones inflamatorias en ambos hemisferios. Hasta el año 2005, alrededor de 200 casos fueron reportados en la literatura, encontrándose participación bihemisférica solo en 924.
En la actualidad existe una amplia gama de estrategias terapéuticas disponibles, tales como bolos de metilprednisolona por vía intravenosa, inmunosupresores, inmunoglobulinas por vía intravenosa (IVIG), plasmaféresis, ciclofosfamida y rituximab. Se reserva el tratamiento quirúrgico para el control de las convulsiones en niños y en adultos refractarios a otras medidas24,28.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


