










To analyze clinical characteristics, survival and causes of death of patients diagnosed with autoimmune inflammatory myositis in the REMICAM registry from the Society of Rheumatology in the Community of Madrid (SORCOM).
MethodsMulticenter cohort of patients diagnosed with autoimmune inflammatory myopathy with follow-up between January 1980 and December 2014. A total of 313 variables concerning demographic, clinical and morbidity data were collected, and a comparison was performed between clinical subgroups.
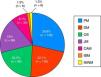
ResultsA total of 479 patients were recruited from 12 centers with 14% of patients lost to follow-up. Seventy-four percent of cases were women, age at diagnosis of 44±23 years and a mean follow-up period of 10±8 years. The most frequent clinical subgroups were primary myositis (PM 29%, DM 22%), followed by overlap myositis (20.5%), juvenile myositis (18%), myositis associated with cancer (8%), immune-mediated necrotizing myositis (1%) and inclusion body myositis (1%). During the follow-up period, a total of 114 deaths (28%) were registered, the main causes being cancer (24%), infections (23%) and cardiovascular events (21%).
ConclusionsA total of 479 patients were recruited in the REMICAM registry of inflammatory myopathies. Including sociodemographic, clinical and prognostic information, it represents the largest Spanish multicenter registry to date in rheumatology, and constitutes an important source for conducting further substudies.
Describir las características clínicas, mortalidad y causas de muerte de una serie de pacientes diagnosticados de miositis inflamatoria idiopática del registro REMICAM de la Sociedad de Reumatología de la Comunidad de Madrid (SORCOM).
MétodosEstudio descriptivo retrospectivo multicéntrico de una cohorte de pacientes con diagnóstico de miositis inflamatoria idiopática en seguimiento en servicios de reumatología de hospitales de la Comunidad de Madrid entre enero de 1980 y diciembre de 2014. Se han recogido hasta un total de 313 variables acerca de aspectos demográficos, clínicos y de morbimortalidad, y se ha realizado una comparación entre subgrupos clínicos.
ResultadosSe han reclutado 479 pacientes procedentes de 12 centros, con un 14% de pérdidas durante el periodo de seguimiento. El 74% de los casos eran mujeres, una edad al diagnóstico de 44±23 años, y una media de seguimiento de 10±8 años. Los subgrupos clínicos más frecuentes fueron las formas primarias (PM 29%, DM 22%), seguidas de síndrome de solapamiento (20,5%), miopatías juveniles (18%), miopatías asociadas a cáncer (8%), miopatías necrosantes inmunomediadas (1%) y miositis por cuerpos de inclusión (1%). Durante el periodo de seguimiento se produjeron un total de 114 fallecimientos (28%), siendo las principales causas el cáncer (24%), las infecciones (23%) y los eventos cardiovasculares (21%).
ConclusionesEn el registro REMICAM de miopatías inflamatorias de la Comunidad de Madrid se han reclutado 479 casos de miositis inflamatoria idiopática con datos sociodemográficos, clínicos y pronósticos, suponiendo el mayor registro multicéntrico español en el ámbito de la Reumatología hasta la fecha, y constituyendo una fuente importante para la realización de posteriores subestudios.
Idiopathic inflammatory myopathies (IIM) comprise a heterogenic group of systemic diseases that are characterized by nonsuppurative inflammation of skeletal muscle and progressive muscle weakness, occasionally accompanied by systemic manifestations. It is considered a rare disease with a worldwide mean incidence between 19cases/million population per year.1 This varies depending on the geographic region, the research methods utilized and the classification criteria applied. Before the use of steroids and immunosuppressive therapy, the rates of mortality were high.2,3 The generalized utilization of these treatments, as well as early diagnosis, resulted in a significant improvement in survival.4–6 The clinical manifestations of IIM, as well as its course and prognosis, are extremely heterogeneous, which together with its low prevalence and the lack of multicenter studies, makes the study of the disease a difficult undertaking.
To date, multicenter studies of IIM in the rheumatology setting have not been performed either in the Community of Madrid or in the rest of Spain. For this reason, and with the support of the Society of Rheumatology of the Community of Madrid (SORCOM), we proposed the creation of a group for the Registry of Inflammatory Myopathies in the autonomous community of Madrid (REMICAM) for the cross-sectional registry of patients, the period of inclusion of which has concluded. The availability of a multicenter observational registry of patients with IIM could enable us to envision the reality of this disease in the Community of Madrid, as well as to determine the rates of morbidity and mortality and compare subgroups of patients, in a disease whose rarity limits the possibility of obtaining significant data.
Patients and MethodsStudy ObjectivesThe main objectives of the REMICAM are to describe and characterize IIM patients, who usually managed in the rheumatology departments of the Community of Madrid, for the purpose of:
- 1.
Describing the sociodemographic and clinical characteristics
- 2.
Studying the rate of comorbidity and the cumulative incidence, and possible differences among distinct types of myopathies
- 3.
Calculating the mortality rate in the overall series and by clinical subgroups
As secondary objectives, the registry proposes a multicenter collaborative study to subsequently examine specific aspects of autoimmune inflammatory myopathies.
Study DesignRetrospective multicenter registry of patients with IIM from units or departments of rheumatology in the Community of Madrid, using a hospital-based cross-sectional registry, recording retrospective information on clinical data, mortality and cause of death.
Patient SelectionThrough SORCOM, the invitation to participate in the present study was sent to the different departments or units of rheumatology of public hospitals in the Community of Madrid, as well as the rheumatology department of Hospital Madrid Norte in Sanchinarro, a borough of Madrid. We included unselected consecutive patients with a diagnosis of IIM (dermatomyositis, polymyositis, inclusion body myositis and immune-mediated necrotizing myositis), who had undergone follow-up in the above departments at some time during the period between January 1980 and December 2014, without taking into account their age at onset of the process. The patients ultimately selected met the criteria of Bohan and Peter7,8 and/or Tanimoto's classification criteria,9 and we excluded toxic and infectious myopathies and myopathies secondary to neuromuscular disease. The patients were classified into 7 subgroups: idiopathic polymyositis (PM), idiopathic dermatomyositis (DM), juvenile myositis (JM), IIM associated with another connective tissue disease (overlap syndrome), cancer-associated IIM, inclusion body myositis10 and immune-mediated necrotizing myositis.11 Patients with overlap syndrome had to meet criteria for IIM and criteria for the following connective tissue diseases: rheumatoid arthritis,12 systemic sclerosis,13 systemic lupus erythematosus,14 mixed connective tissue disease15 or Sjögren's syndrome.16 We defined myositis as being cancer-associated in those cases in which cancer was diagnosed during the 3 years prior to or after the development of the myopathy, similar to the approach used in previous studies.17 For the comparison of the different subgroups, we only took into account the first 5 of those mentioned above, given the low prevalence of inclusion body myositis and immune-mediated necrotizing myositis.
VariablesIn all, 313 variables were included for each patient, and were grouped in terms of their relation to sociodemographic characteristics, classification, mortality, comorbidity, clinical manifestations and analytical data, as well as information regarding treatments. All of these data were retrieved from the medical history of each patient. Exhaustive monitoring for inconsistencies in the data was performed by an external company.
The sociodemographic data included information on sex, race, age at disease onset, age at the last visit (age of the patient at the time of the last visit available or age at the time of death), disease duration (time since the diagnosis and the last visit).
General symptoms included weight loss >10% or fever not explained by causes other than the rheumatic disease. Skin manifestations included Gottron's papules, heliotrope rash, Gottron's sign, cutaneous vasculitis (established by a compatible biopsy), photosensitivity, mechanic's hands, nonspecific pruritus, skin ulcers, ischemic ulcers in fingertips, periungual erythema and/or macroscopic dilatation of nailfold capillaries, hand edema and sclerodactyly. The hematologic manifestations included anemia, leukopenia or thrombocytopenia attributed to the disease, after ruling out drug-related and infectious causes, among others. We took into account gastrointestinal manifestations such as: dysphagia, gastroesophageal reflux (defined as suggestive clinical signs and upper gastrointestinal tract endoscopy and/or pH-metry and/or esophageal manometry showing evidence of gastrointestinal tract dysmotility, after ruling out other causes), upper/lower gastrointestinal bleeding attributed to the disease (defined as hematemesis, melena or rectal bleeding, after ruling out other causes) and diarrhea or constipation attributed to the disease (defined by compatible clinical signs, after ruling out other causes). As cardiovascular disease, we included: arterial or venous or pulmonary thromboembolic disease, ischemic cardiomyopathy, arrhythmias, stroke and pulmonary hypertension. Pulmonary hypertension was defined by echocardiogram (estimated systolic pulmonary artery pressure [sPAP] ≥40mmHg) and/or cardiac catheterization (measured sPAP ≥25mmHg). Interstitial lung disease was diagnosed on the basis of compatible clinical signs (recent onset dyspnea on exertion and/or dry cough and/or fever with no other origin) and diagnostic chest radiographic images, high-resolution computed tomography of the lungs and/or compatible lung biopsy, and/or compatible respiratory function tests, once other causes had been ruled out. Interstitial lung disease was classified, when possible, depending on the result of the lung biopsy and/or the radiologic image and, in the case of disagreement, a consensus was reached between the radiologist and the pathologist to define the following subtypes: nonspecific interstitial pneumonia, usual interstitial pneumonia, bronchiolitis obliterans organizing pneumonia, diffuse alveolar damage and cryptogenic pneumonia. An infection was considered to be serious if it required hospital admission or led to death.
The cause of death of patients in routine follow-up in their referral hospital was recorded in terms of the data on the medical history or the death certificate. When a patient was lost to follow-up, the attempt was made to make contact by telephone to determine the status of the patient, as well as the cause and date of death. The causes of death were classified according to 5 subgroups depending on the underlying cause: infections, cardiovascular event (arrhythmia, ischemic cardiomyopathy, acute ischemic or hemorrhagic stroke, pulmonary hypertension, heart failure and pulmonary thromboembolism), cancer, interstitial lung disease and miscellaneous.
Statistical AnalysisA significance level α of .05 was assumed for all of the statistical analyses. Sociodemographic factors, classification subgroups, comorbidity and survival were assessed by basic descriptive statistical analysis. For the description of the sample, we used central tendency measures (mean and median) and dispersion (standard deviation and interquartile range), as well as frequency tables and percentage distribution for continuous and categorical variables, respectively. The comparison of the distinct types of myopathies was done using mean difference tests, with analysis of variance, comparison of proportions by means of chi-square test, and Fisher's exact test in cases of cell sizes less than 5. All of the analyses were performed with Stata 12.
ResultsThe sample was comprised of 479 patients from rheumatology departments or units from 12 hospitals of the Community of Madrid (Table 1). Seventy patients (14.6%) had been lost to follow-up at the time of inclusion in the registry.
Distribution of Patients by Hospital.
| Hospital (No.=479) | n (%) |
|---|---|
| La Paz | 57 (11.9) |
| Ramón y Cajal | 38 (7.9) |
| La Princesa | 14 (2.9) |
| Puerta de Hierro | 14 (2.9) |
| Fundación Alcorcón | 5 (1) |
| Sanchinarro | 14 (2.9) |
| Gregorio Marañón | 169 (35.3) |
| Infanta Sofía | 6 (1.3) |
| Príncipe de Asturias | 11 (2.3) |
| Infanta Leonor | 4 (0.8) |
| Doce de Octubre | 134 (28) |
| Niño Jesús | 13 (2.7) |
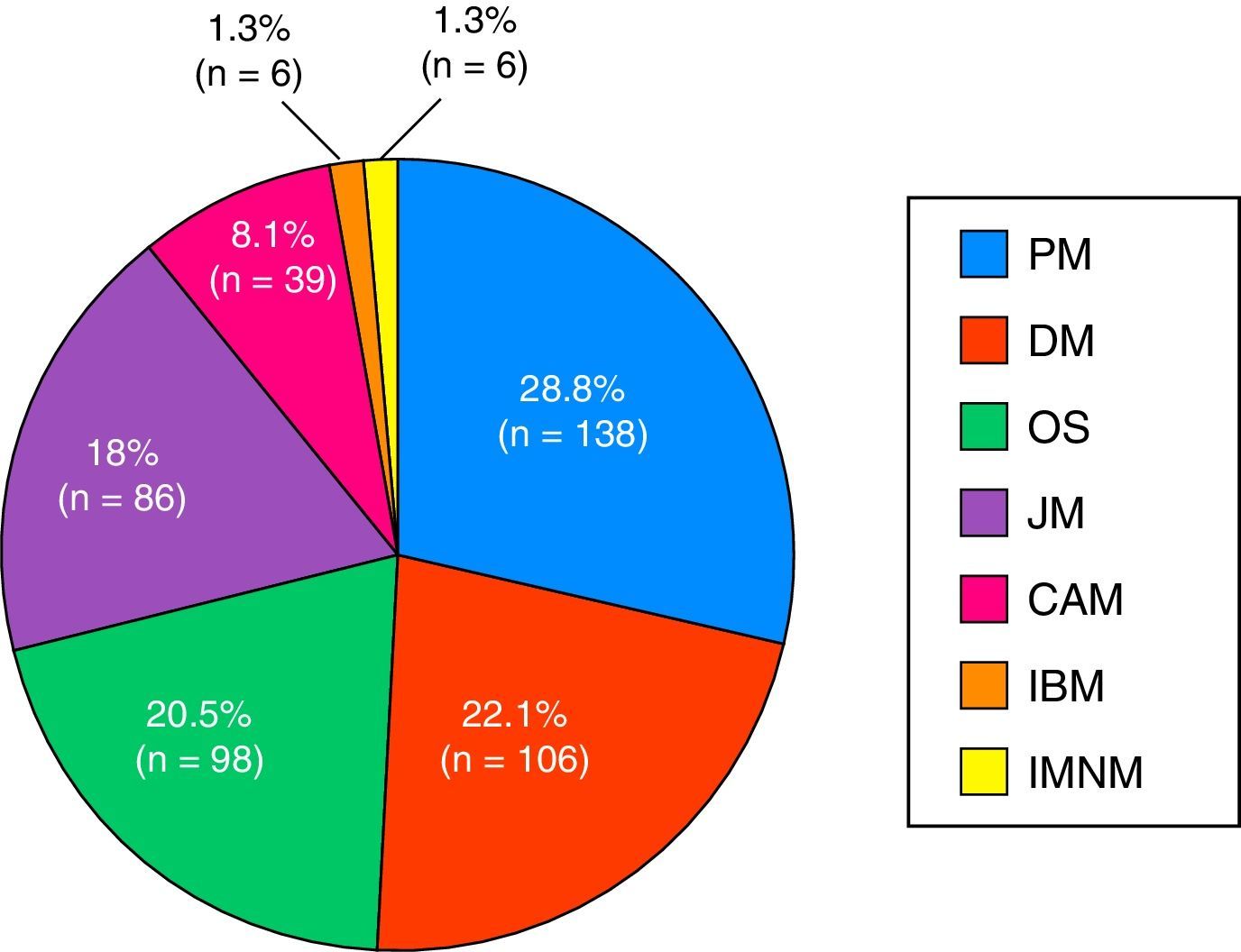
The most widespread myopathy subgroups were the primary forms (PM and DM), followed by overlap syndrome, juvenile myositis and cancer-associated myositis. Moreover, there were 6 cases of inclusion body myositis and 6 patients with immune-mediated necrotizing myositis (Fig. 1). Of the 6 patients with immune-mediated necrotizing myositis, only 1 had undergone treatment with statins and 3 had not. It was not possible to obtain this information for the other 2 patients. The connective tissue disease most frequently associated with IIM in overlap syndrome was mixed connective tissue disease, followed by systemic sclerosis, systemic lupus erythematosus, Sjögren's syndrome and rheumatoid arthritis (Fig. 2). In all, 99.6% met the criteria of Bohan and Peter, 97.3% Tanimoto's criteria, and 96.9% both. According to the classification criteria proposed by Bohan and Peter, 99.6% of the patients would have a possible disease, 94.1% a probable disease and 67.4% a definite disease. All of the patients but 1 with a diagnosis of overlap syndrome had a muscle biopsy compatible with myositis or electromyographic study with a typical myopathic pattern. That patient was diagnosed with systemic sclerosis and had muscle weakness and a baseline creatine kinase (CK) level of 748IU/L, but later improved with treatment.
Most of the patients in our series were women (74.1%) and Caucasian (93.5%). The remaining sociodemographic data are shown in Table 2. With regard to clinical manifestations, there was a marked prevalence of interstitial lung disease (29.9%), mainly usual interstitial pneumonia (28.2%) and nonspecific interstitial pneumonia (23.4%), followed by bronchiolitis obliterans organizing pneumonia (4%), diffuse alveolar damage (2.4%) and cryptogenic pneumonia (0.8%); it was not possible to identify the subtype of the remainder of the patients. The CK levels in 11.6% of the patients were higher than normal at the end of follow-up, and the follow-up period was longer in patients with normal CK levels (10.3±8.5 years vs 5.9±5.2 years; P<.001).
Differential Clinical Characteristics According to Clinical Subgroup, n (%).
| PM (No.=137) | DM (No.=107) | JM (No.=86) | OS (No.=98) | CAM (No.=39) | Total | |
|---|---|---|---|---|---|---|
| Age at diagnosis*** | 54±17 | 50±16 | 8±4 | 45±17 | 62±11 | 43.7±22.6 |
| Age at end of follow-up*** | 62±17 | 60±15 | 17±10 | 55±17 | 62±11 | 52.5±22.6 |
| Time since diagnosis*** | 8±7 | 11±9 | 8±8 | 13±9 | 5±7 | 9.7±8.3 |
| Female sex** | 96 (70.1) | 86 (80.4) | 61 (70.9) | 84 (85.7) | 21 (53.8) | 74.1 |
| Arthritis*** | 47 (34.6) | 50 (48.1) | 24 (28.2) | 64 (66) | 11 (29.7) | 196 (42.7) |
| Skin manifestations*** | 29 (21.2) | 107 (100) | 79 (91.9) | 63 (64.3) | 30 (76.9) | 308 (66) |
| Typical signs of DM*** | 0 | 107 (100) | 73 (84.9) | 22 (22.4) | 26 (66.7) | 225 (48.8) |
| Hematologic manifestations*** | 28 (20.6) | 26 (24.3) | 16 (18.6) | 52 (53.6) | 11 (28.9) | 133 (28.7) |
| ILD*** | 48 (35) | 33 (30.8) | 2 (2.3) | 47 (48) | 8 (21.1) | 138 (29.6) |
| Gastrointestinal manifestations** | 33 (24.1) | 37 (34.6) | 21 (24.4) | 44 (44.9) | 18 (46.2) | 153 (32.8) |
| Dysphagia** | 25 (18.2) | 28 (26.4) | 19 (22.1) | 41 (41.8) | 11 (28.2) | 124 (26.6) |
| General manifestations* | 41 (30.6) | 46 (44.7) | 25 (31.6) | 50 (51) | 16 (45.7) | 178 (39.6) |
| Cardiac manifestations** | 34 (24.8) | 19 (17.8) | 6 (7) | 26 (26.5) | 9 (23.1) | 94 (20.1) |
| Pulmonary hypertension** | 10 (7.4) | 11 (10.3) | 1 (1.2) | 15 (15.3) | 1 (2.6) | 38 (8.2) |
| Muscle involvement | 137 (100) | 107 (100) | 85 (98.8) | 98 (100) | 39 (100) | 466 (99.8) |
| Muscle weakness | 129 (94.2) | 104 (97.2) | 80 (93) | 95 (96.9) | 38 (97.4) | 446 (95.5) |
| Baseline CK level (IU/L) | 2911±4556 | 1955±3557 | 2264±3512 | 1324±2478 | 1900±3538 | 2161±3723 |
| Final CK level (IU/L)* | 305±655 | 122±153 | 199±457 | 142±301 | 87±96 | 192±445 |
| Calcinosis*** | 3 (2.2) | 9 (8.6) | 27 (31.4) | 10 (10.2) | 0 | 49 (10.6) |
| Raynaud's*** | 23 (16.9) | 21 (19.8) | 12 (14) | 64 (65.3) | 13 (33.3) | 133 (28.6) |
| Serious infection*** | 36 (26.7) | 22 (21) | 3 (3.8) | 38 (41.3) | 12 (34.3) | 111 (24.9) |
| Cancer*** | 16 (11.7) | 8 (7.5) | 1 (1.2) | 6 (6.1) | 39 (100) | 70 (15) |
| Death*** | 32 (26) | 19 (21.3) | 4 (6.2) | 30 (37) | 28 (71.8) | 113 (28.5) |
CAM, cancer-associated myositis; CK, creatine kinase; DM, idiopathic dermatomyositis; ILD, interstitial lung disease; JM, juvenile myositis; PM, idiopathic polymyositis; OS, overlap syndrome.
Ages, time since diagnosis and CK levels are expressed as mean±standard deviation; the remaining variables are expressed as number and percentage (in parenthesis).
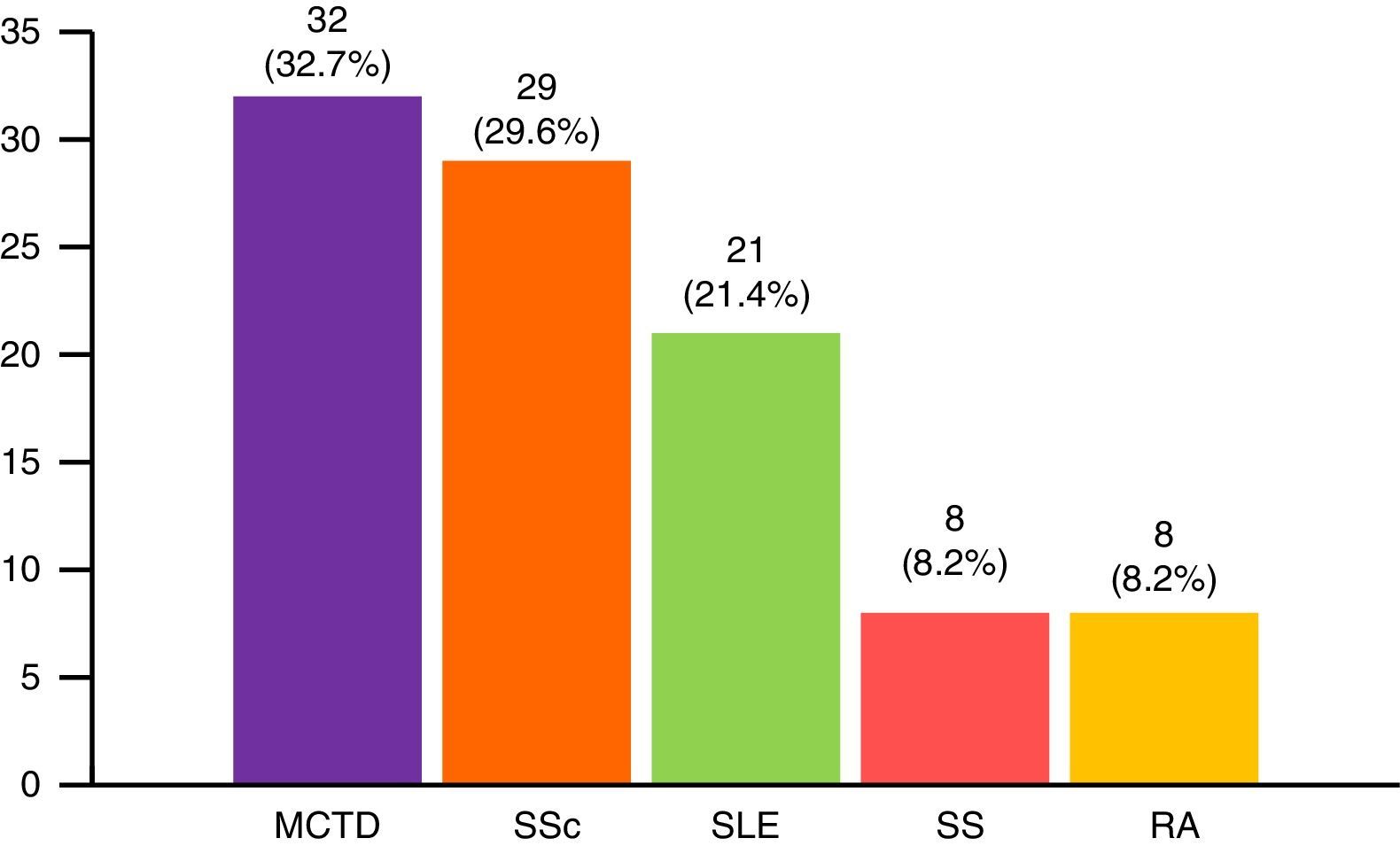
Patients with DM experienced a greater number of general symptoms when compared with those with PM, and mechanic's hands and typical skin manifestations were more frequent. Juvenile myositis was characterized by more frequent typical skin manifestations than was observed in dermatomyositis, but Raynaud's phenomenon, extramuscular manifestations, malignant neoplasms and serious infections were less common. On the other hand, overlap syndrome was more frequently accompanied by arthritis, Raynaud's phenomenon, hematologic manifestations, interstitial lung disease, and systemic, neuropsychiatric and renal manifestations. Paraneoplastic myositis had a shorter follow-up and the patients had lower CK levels at the end of follow-up when compared to the other subgroups. The most common myositis specific antibodies were anti-Jo-1 (77 cases), followed by anti-RNP, anti-Mi-2, anti-PM-Scl and others less frequent (Fig. 3). The remaining clinical manifestations are shown in Table 2.
Comorbidity and Complications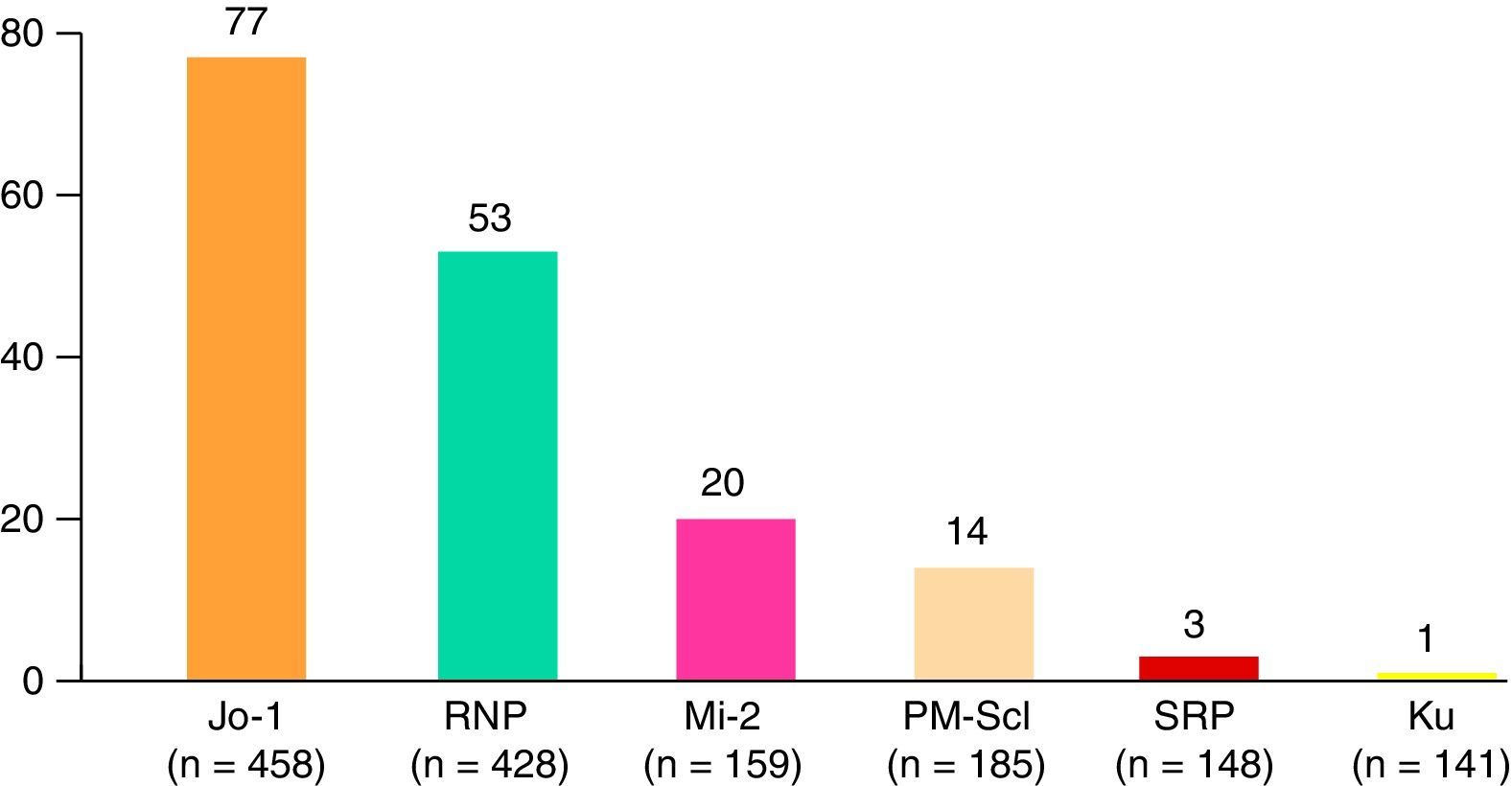
Overall, 54.6% of the patients had some cardiovascular risk factor, mostly dyslipidemia, hypertension, diabetes mellitus and active tobacco use, and 22.3% of the cases were complicated by one or more cardiovascular events (Table 3).
Comorbidity and Complications.
| Comorbidity | n (%) |
|---|---|
| Cardiovascular disease | 104 (22.3) |
| Peripheral vascular disease | 26 (5.6) |
| Ischemic cardiomyopathy | 28 (6) |
| Stroke | 23 (4.9) |
| Pulmonary hypertension | 38 (7.9) |
| Heart failure | 45 (9.6) |
| Hypertension | 146 (31.1) |
| Diabetes mellitus | 58 (12.4) |
| Dyslipidemia | 149 (31.8) |
| Active tobacco use | 64 (19.6) |
| Cardiovascular risk factors (hypertension, diabetes, dyslipidemia, tobacco use) | 261 (54.6) |
| Number of cardiovascular risk factors | |
| One | 141 (29.9) |
| Two | 85 (18) |
| Three | 34 (7.2) |
| Four | 1 (0.2) |
| Pulmonary disease | 40 (8.5) |
| Pleuritis | 13 (2.7) |
| COPD | 27 (5.8) |
| Complications | |
| Serious infections | 113 (24.7) |
| Cancer | 70 (14.6) |
| Death | 114 (27.9) |
COPD, chronic obstructive pulmonary disease.
In all, 25% of the patients had complications in the form of serious infections, mainly lower respiratory tract infections (63%), followed by urinary tract (14%), gastrointestinal (10%) and others that were less common. In 30% of the cases of serious infections, the outcome was death. Serious infections developed more frequently in patients with overlap syndrome and myopathy associated with a malignant neoplasm. With regard to cancer-associated myositis, the tumors most frequently involved were lung and skin cancer (8 cases each), lymphoma (n=7) and breast cancer (n=6). In 2 cases, the origin of the cancer had not been determined.
Death and the CausesDuring the study period, there were 114 deaths, representing 28% of the analysis sample. The major causes were cancer, infections and cardiovascular events (Fig. 4). Patients with cancer-associated myositis had the worst prognosis, followed by overlap syndrome, whereas the patients with juvenile myositis had the best prognosis.
Treatment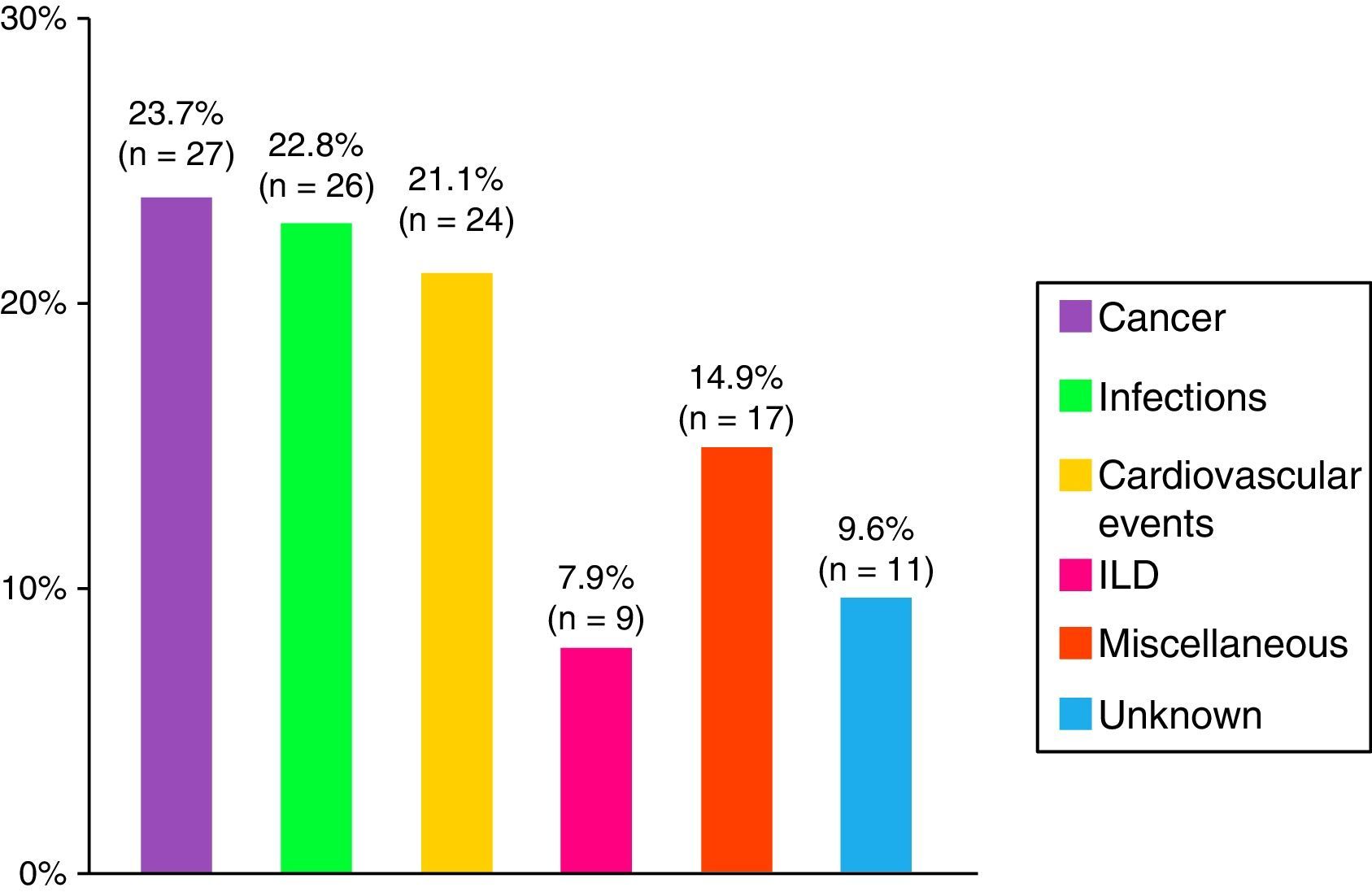
With regard to treatment, a substantial number of the patients had received, at some time, glucocorticoids, either intravenously (15%) or orally (99%), and half of them (50.3%) still required oral steroids when they visited their physician for the last time. They were taking a dose of less than 30mg/day in 95% of the cases at the end of follow-up. In all, 13.2% of the patients required more than 1 cycle of oral steroids over the course of the disease. Immunosuppressive agents were utilized in 74.1% of those patients, 30.5% of whom required the use of 2 or more drugs over the course of the disease (with an average of 2.4±0.9 immunosuppressive drugs in those cases). The nonbiologic therapies most widely utilized, in cumulative percentage, were methotrexate (48%), azathioprine (40%), hydroxychloroquine (16%) and cyclophosphamide (9%). In all, 9% were treated with intravenous immunoglobulin and 12% received biological therapy at some time (the drug most frequently used was rituximab in 9% of the cases) (Fig. 5).
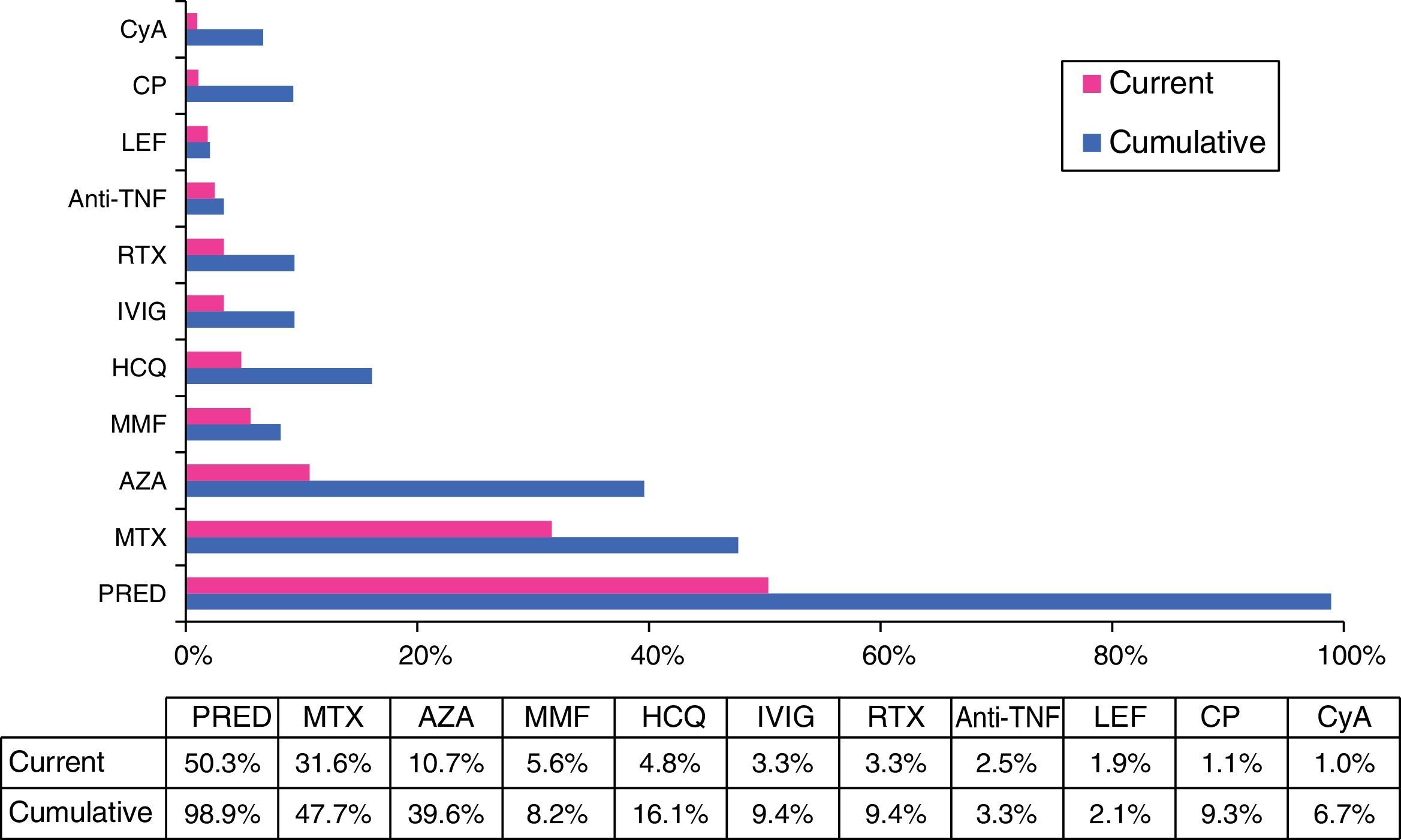
Cumulative and current treatment.
Anti-TNFα, anti-tumor necrosis factor α; AZA, azathioprine; CP, cyclophosphamide; CyA, cyclosporine A; HCQ, hydroxychloroquine; IVIG, intravenous immunoglobulins; LEF, leflunomide; MMF, mycophenolate mofetil; MTX, methotrexate; PRED, prednisone; RTX, rituximab.
This is the first multicenter registry of inflammatory myopathies in the Community of Madrid, and it provides the most extensive compilation of data up to now. The registry was created for the purpose of retrospectively analyzing the clinical characteristics, comorbidities and complications of autoimmune inflammatory myopathies in a multicenter study of 12 hospitals in the Community of Madrid. We compared the clinical characteristics, analytical findings and prognosis in the different subgroups of autoimmune inflammatory myopathies.
The criteria of Tanimoto,9 published in 1995, have a greater specificity than those of Bohan and Peter.18,19 The only 2 cases that did not meet the criteria of Bohan and Peter, did conform to Tanimoto's criteria, and corresponded to 2 patients with antisynthetase syndrome with elevated muscle enzymes, arthritis, general symptoms and interstitial lung disease in both cases, and mechanic's hands in one.
Idiopathic inflammatory myopathies appear to be associated with a number of connective tissue diseases, and are reported to have a highly variable incidence, between 7% and 60%, depending on the series.6,20–23 In the present study, we employed widely accepted classification criteria, to conclude that the patients actually did have a connective tissue disease associated with inflammatory myopathy.12–16 In all, 21% of the cases were classified as overlap syndrome, a prevalence that is very similar to that of a Hungarian series,20 although greater than that of another Spanish series from a single hospital.6 Some of the patients from that series were included in the present registry. The differences observed in terms of the higher proportion of patients with overlap syndrome in the REMICAM with respect to the other Spanish series could be due to the development of new manifestations in those patients that enabled the diagnosis of other associated connective tissue diseases, as was proposed by other authors,24 or to differences in the characteristics of the other patients included.
Only 11.6% of the patients had CK levels over the normal range at the end of follow-up, a fact that may give an approximate idea of the patients in whom activity persisted. Nevertheless, the addition of other data, such as those proposed by the Myositis Outcome Assessment Collaborative Study Group (IMACS) and the Paediatric Rheumatology International Trials Organisation (PRINTO) for the definition of disease activity, such as measuring muscle strength, global assessment of the disease by the patient/parents and the physician using visual analogue scales, questionnaires on physical function and other data, could have given a more precise idea of disease activity.25,26 However, retrospective study limits the collection of these data, as well as data to evaluate disease relapse.
As was expected with respect to the clinical differences among the subgroups, patients with overlap syndrome had a different disease, with symptoms characteristic of other conditions, mainly systemic sclerosis or systemic lupus erythematosus, similar to the findings in other series.6 Concerning associated comorbidity, it is noteworthy that early half of the patients had one or more cardiovascular risk factors, dyslipidemia being the most prevalent, followed by hypertension.
The incidence of cancer in myopathy varies widely from one report to another,27,28 depending mostly on the definition of the subgroup of cancer-associated myositis, as well as the age and sex of the patients, and the characteristics of the series. The REMICAM cohort had a lower incidence than the majority of the series in the literature. This is probably due to the definition of cancer-associated myositis, as well as to the inclusion of patients with overlap syndrome and the lower percentage of cases of DM, which has classically been related to a higher risk of cancer.6,17
Overall mortality in REMICAM was 28.5%, similar to that of another Spanish and Asian series, but higher than that of other patient populations.6,29 The most frequent cause of death was cancer, closely followed by infections and cardiovascular events, as has been reported by others.30,31 In the majority of the series, mortality depends on the inclusion criteria utilized, on the associated diseases, the duration of patient follow-up and, decisively, on the inclusion or exclusion of cancer-associated myositis, which has a negative effect on the prognosis. Thus, the higher survival of in some series could be due to the exclusion of cancer-associated myositis, to the higher proportion of cases of juvenile disease, to the younger age of the patients and to the high rate of lost studies.4,5
This report is susceptible to selection biases, mainly related to the method of selecting the centers. For example, it could be that the patients in the hospitals interested in participating were in poorer health or were studied more extensively. However, this bias is limited by the fact that the present study was also open to specialty centers, where there would likely be more benign cases, as well as to the fact that IIM are long-standing diseases and are associated with a high mortality rate, which makes it probable that the majority of the patient were referred from tertiary centers at some time during the course of the disease.
On the other hand, the retrospective design of the study impedes controlling the quality of the data, allows the introduction of measuring errors and accessibility to information on important confounders. However, the low proportion of the loss of patients and the exhaustive monitoring for inconsistencies on the part of the external company makes it little likely that this limitation had an influential effect on the quality of the results.
To date, REMICAM is the largest registry of patients with myopathy in the Community of Madrid, and in the rest of Spain, in the rheumatology setting, based on a detailed study of clinical characteristics, morbidity, mortality and clinical subgroups. The performance of further substudies will enable us to expand our knowledge on the course of this disease.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
FundingThe work for the present study was partially financed by a grant from SORCOM-MSD.
Conflicts of InterestThe authors declare they have no conflicts of interest.
To the Board of Directors of SORCOM, and especially to Santos Castañeda and Ana Cruz, for their unconditional support and commitment to this project; to Loreto Carmona for her advice and guidance in the methodology; and to the SER for its logistic support for the meetings of our group.
Please cite this article as: Nuño L, Joven B, Carreira P, Maldonado V, Larena C, Llorente I, et al. Registro de pacientes con miopatía inflamatoria de la Sociedad Madrileña de Reumatología: análisis descriptivo. Reumatol Clin. 2017;13:331–337.


