

C1q deficiency is a rare autosomal recessive genetic condition characterized by mutations in genes C1qA, C1qB, or C1qC which can cause a SLE-like disease. Here, we report the cases of two siblings with C1q deficiency, both of whom had homozygous mutations in the C1QA gene. Both of our patients had NP involvement, and the brother had chilblain lesions. Diagnosis of C1q deficiency was delayed, highlighting the importance of clinical suspicion and genetic testing. This is especially crucial in cases with atypical presentations of SLE and a family history of consanguinity.
La deficiencia de C1q es una condición genética autosómica recesiva poco frecuente, caracterizada por mutaciones en los genes C1qA, C1qB o C1qC, que puede ocasionar una enfermedad semejante al LES. En este informe, presentamos los casos de dos hermanos con deficiencia de C1q, ambos con mutaciones homocigotas en el gen C1QA. Nuestros dos pacientes presentaron compromiso neuropsiquiátrico, y el hermano manifestó lesiones tipo perniosis. El diagnóstico de deficiencia de C1q se retrasó, lo que pone de relieve la importancia de la sospecha clínica y las pruebas genéticas. Esto cobra especial relevancia en casos con manifestaciones atípicas de LES y antecedentes familiares de consanguinidad.
C1q deficiency is a rare autosomal recessive genetic condition characterized by mutations in genes C1qA, C1qB, or C1qC. Clinical features include recurrent infections with encapsulated bacteria and Systemic Lupus Erythematosus (SLE)-like disease.1,2 The onset is earlier than idiopathic SLE, with oral ulcers and discoid rash more frequent. While anti-dsDNA positivity and arthritis are less common.2 Neuropsychiatric (NP) involvement is less common but can be severe.3 Here, we report the cases of two siblings with C1q deficiency, both of which have homozygous mutations in the C1QA gene.
Case 1A 10-year-old male patient initially presented with bilateral lower extremity rash and edema. He was diagnosed with Henoch–Schönlein Purpura (HSP). Seven years later, he developed arthritis in his elbow and had a history of thrombocytopenia, lymphopenia, migratory arthritis, serositis, peritonitis, and oral ulcers. Laboratory tests showed ANA positivity (1:250 speckled) and positive anti-Smith, anti-RNP, and anti-Ro antibodies, while anti-La and anti-dsDNA were negative. His complement levels were normal. He was diagnosed with SLE and given hydroxychloroquine and prednisolone. A renal biopsy indicated membranoproliferative glomerulonephritis.
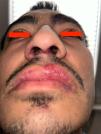
His parents were in a third-degree consanguineous marriage. He had neuropsychiatric symptoms such as attention deficit and mild mental retardation. A skin biopsy revealed IF (−) leukocytoclastic vasculitis. Over nine years, he experienced recurring flares with digital ulcers, malar rash, nasal ulcers, arthritis, oral ulcers, alopecia, chilblain lesions, and photosensitive rashes (Fig. 1). Next-generation sequencing (NGS) identified a homozygous frameshift mutation in the C1QA gene (c.320del p.(Gly107Glufs*175)), confirming C1q deficiency. Of his four siblings, one also has C1q deficiency, while the others are healthy. His treatment included hydroxychloroquine, ramipril, prednisolone, mycophenolate mofetil (MMF), and amlodipine.
Case 2
A 9-year-old female patient presented with abdominal pain, bilateral lower extremity rash, proteinuria, and hematuria and was diagnosed with HSP. Two years later, a renal biopsy revealed C3 glomerulopathy, and a skin biopsy showed leukocytoclastic and urticarial vasculitis. When referred to our clinic, she had positive p-ANCA, RF of 20.2IU/mL, C3 of 1.21g/L, and C4 of 0.68g/L. She was prescribed prednisolone, MMF, and hydroxychloroquine.
Later that year, she developed dysuria, urge incontinence, tremors, and bilateral livedo reticularis. ANA was borderline positive with speckled pattern, anti-dsDNA was slightly elevated, and EMG was normal. In 2023, NGS revealed the same homozygous C1QA mutation as her brother.
DiscussionBoth siblings initially presented with an HSP-like presentation. The male patient later showed SLE features, while the female had a more elusive disease course. Both had ANA positivity, yet neither had anti-dsDNA positivity. NP involvement and vasculitis were observed in both. Although they received various immunosuppressants, diagnosis of C1q deficiency was delayed.
Due to the rarity of C1q deficiency, clinical suspicion is crucial for diagnosis. Patients with C1q deficiency exhibit a wide range of clinical presentations,4 making it essential to consider specific clinical characteristics before a definitive diagnosis. SLE-like disease in C1q deficiency patients manifest at a younger age compared to sporadic lupus. In Indian juvenile SLE patients, 20% had complement deficiency, highlighting the importance of including complement deficiency in differential diagnosis.5 A thorough family history is critical due to autosomal recessive inheritance and frequent consanguinity. Minors with rare syndromes and lupus-like symptoms may indicate C1q deficiency, as the co-occurrence is not unexpected.6 Further studies are needed to distinguish idiopathic SLE patients from those with C1q deficiency.
ConclusionsA high index of suspicion is vital for pediatric patients with connective tissue disease symptoms not meeting classic SLE criteria. Genetic testing, particularly of the complement system, is warranted. Early recognition of C1q deficiency is essential to minimize long-term complications.
Authors’ contributionsKerem Parlar: Conceptualization, data curation, and writing-original draft. Berkay Aktaş: Data curation, and writing-original draft. Sena Ladin Sıcakyüz: Data Curation, and writing-review and editing. Sezgin Şahin: Supervision, and writing-review and editing. Özgür Kasapçopur: Supervision, and writing-review and editing. Serdal Uğurlu: Conceptualization, supervision, and writing-review and editing.
Patient consentWritten informed consent was obtained from the patient for publication of this case report.
Ethical statementEthical approval is not applicable.
Conflict of interestThe authors declare no conflict of interest. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
None.


