





TGF-β is a cytokine with pleiotropic functions in hematopoiesis, angiogenesis, cell proliferation, differentiation, migration and apoptosis. Although its role in rheumatoid arthritis is not well defined, TGF-β activation leads to functional immunomodulatory effects according to environmental conditions. The function of TGF-β in the development of arthritis in murine models has been extensively studied with controversial results. Recent findings point to a non-relevant role for TGF-β in a mice model of collagen-induced arthritis. The study of TGF-β on T-cell responses has shown controversial results as an inhibitor or promoter of the inflammatory response. This paper presents a review of the role of TGF-β in animal models of arthritis.
El TGF-β es una citocina implicada en procesos celulares como hematopoyesis, proliferación, angiogénesis, diferenciación, migración y apoptosis celular. Aunque su papel en la artritis reumatoide no está bien definido, está considerada como una citocina inmunomoduladora según las condiciones del entorno. Numerosos trabajos han tratado de definir el papel del TGF-β en el desarrollo de la artritis murina en diferentes modelos de enfermedad, con resultados discordantes. De hecho, resultados recientemente publicados indican que TGF-β no desempeña un papel relevante en el modelo murino de artritis inducida con colágeno. Su implicación en la diferenciación y la funcionalidad de las diferentes poblaciones de células T también ha mostrado resultados dispares sobre su papel como inhibidor o promotor de la respuesta inflamatoria. En este trabajo se presenta una revisión sobre el papel de TGF-β en modelos animales de artritis.
Rheumatoid arthritis (RA) is an autoimmune disease of unknown etiology, characterized by chronic inflammation that affects diarthrodial joints, has a progressive course and causes joint functional disability, deformity and, ultimately, a decreased quality of life and reduced life expectancy. Its pathogenesis is characterized by an alteration of cellular and humoral immunity, and impaired resident cell components of the connective tissue of the synovial membrane (SM), which behave in a pseudotumoral fashion, invading and destroying adjacent tissue. The first manifestations of RA joint inflammatory response appear to be due to microvascular changes and an increase in the number of cells or synovial lining hyperplasia. These changes are accompanied by an altered regulation of cytokines, an increase in the number of fibroblasts and excess proliferation of inflammatory cells,1 mainly macrophages and lymphocytes, tissue destruction and angiogenesis.2 Rheumatoid mesenchymal fibroblasts secrete proinflammatory cytokines and angiogenic growth factors which recruit synovial cells to the synovial space.3,4 By microarray analysis two subsets of fibroblasts have been identified in the rheumatoid SMIn, the inflamed tissue where there is an overexpression of a cell subtype that synthesizes transforming growth factor-beta (TGF–β) and Activin-A inducible gene, both characteristic of myofibroblasts. The other subgroup expresses genes regulated by insulin-like growth factor (IGF) and appears in less inflamed synovial membranes.5 Activated macrophages are the major producers of inflammatory cytokines, including tumor necrosis factor-alpha (TNF–α). This, in turn, induce the release of other inflammatory cytokines such as interleukin (IL)-1, IL-6 or IL-8, involved in angiogenesis and the proliferation of the synovial fibroblasts and the production of factor platelet-derived growth (PDGF), fibroblast growth factor (FGF) and TGF–β. The sustained activation of these cell types leads to RA's structural alterations. Synovial macrophages, along with SF, play an important role in the tissue destruction characteristic of RA and are the main source of production of metalloproteinases (MMP). This phenomenon is directly regulated by proinflammatory cytokines such as IL-1, TNF–α, and TGF–β secreted in turn by resident macrophages in the synovium. Neutrophils are the most abundant cell type in the synovial fluid and capable of synthesizing TGF–β and IL-8, essential mediators in their own recruitment, establishing an autocrine circuit. In addition to cellular components, soluble factors of the rheumatoid synovial as well as certain proinflammatory cytokines (TNF–α, IL-1, IL-6, IL-17) are involved in activating osteoclastogenesis directly through osteoclast precursors.6,7 It has recently been discovered that the osteoclasts secrete IL-10, TGF–β and IL-6 and may act as antigen presenting cells and activate T CD8+ and CD4+ cells.8
Angiogenesis is a remarkably active process in RA, especially in early stages and is regulated by various pro-angiogenic mediators such as TGF–β, angiopoietin, placenta growth factor, FGF, and vascular growth factor (VEGF). These factors activate endothelial cells and induce the production of proteolytic enzymes that degrade the basement membrane and perivascular extracellular matrix.
In the rheumatoid synovium the expression of 4 growth factors has been described, TGF–β, PDGF, FGF and IGF.9,10 TGF-β is a homodimeric protein involved in the control of many biological processes such as angiogenesis, cell proliferation, differentiation, migration and apoptosis.11 This cytokine has a structure consisting of two disulfide-linked subunits, forming a homodimer of 25kDa. In mammals there are three isoforms of TGF–β (β TGF-1, TGF-2 and TGF–β 3) with similar functions but expressed in different tissues. TGF-β 3 has been detected mainly in cells of mesenchymal origin, indicating a different role of TGF–β 1 and 2. Lymphoid cells mainly produce the TGF–β 1 isoform, hence giving it a greater role in the immune system,12 particularly in the control of proliferation, activation and differentiation of T cells. So far, 3 receptors have been described, TGF–β (TGF–β RI, TGF-RII and TGF–β RIII), but the first 2 are the mediators of the biological responses of TGF–β 1.13
Role of Transforming Growth Factor-Beta in Rheumatoid ArthritisThe role of TGF–β has been studied in various models of disease, such as embryonic defects, cancer, autoimmune diseases, atherosclerosis, hypertension, osteoporosis and14 fibrosing and inflammatory diseases. Numerous studies in fibrosis models have demonstrated the beneficial effect of blocking TGF–β15,16 with antibody-specific anti-TGF-β antisense oligonucleotides,17,18 or synthetic peptides,19–21 demonstrating the role of TGF–β as a profibrotic agent. Other studies have shown that in late stages of tumor progression TGF–β promotes tumorigenesis and induces changes in the differentiation of epithelial tumor cells, a phenomenon known as epithelial–mesenchymal transdifferentiation.22
Genetically modified animal models demonstrate the modulating effect of TGF–β on the immune response. Mice deficient in TGF–β 2 and TGF–β 3 have some defects that are lethal in embryonic development. However, mutations of TGF–β 1 gives rise to serious inflammatory disorders and multiorgan tissue necrosis which rapidly generates prenatal lethality (between 3 and 5 weeks old).12 Also, the functional absence of TGF–β 1 causes an accumulation of cells and pro-inflammatory cytokines (TNF–α, IFN-γ, IL-1 β) in the lymphoid organs, increased production of antigens of the major histocompatibility complex class I and II and a deficit in the proliferation of hematopoietic and endothelial cells.23 Histologically, these mice showed massive infiltration of lymphocytes and macrophages, mainly in the lungs and heart, but are also found in muscle, liver, pancreas and brain, among other organs.24 These mice also exhibit anti-dsDNA, anti-ssDNA and glomerular deposition of immune antibodies. Other experiments have shown that mice transgenic for TGF–β RII (dnTRII β, which express TGF–β RII kinase with an incomplete intracellular domain under a specific promoter for T cells and, therefore, no signaling through the receptor) showed increased susceptibility to arthritis in comparison with WT phenotype mice, accompanied by an increase in the production of TNF-α and IFN–γ T cells in lymphoid nodes.25
Unlike what happens in other diseases where the role of TGF–β is well defined, studies in models of arthritis have exhibited very differing results (Table 1). Multiple studies have reported that intraperitoneal administration of TGF–β 1 will reduce the incidence and severity of arthritis in mice with collagen-induced arthritis (CIA), especially if the injection is made in late stages of the disease.26,27 In rats with arthritis due to Streptococcus (SCW) the intraperitoneal administration of TGF–β in the acute phase suppresses the development of arthritis.28 The same applies with the administration of TGF–β intramuscularly,29 but this is only seen if performed at the peak of inflammation. A very recent study has shown that mesenchymal stem cells derived from bone marrow and induced with TGF–β decrease the incidence and severity of arthritis in CIA mice with established disease. Also, they increase the proportion of Foxp3/IL-17 in the spleen and the peritoneal cavity, and reduce the production of pro-inflammatory cytokines.30 Another study in CIA mice showed a significant increase of TGF-β 1 and TGF–β 2 in the joint,31 particularly during the remission phase, indicating that both cytokines are involved in regulating the activity of the disease. In this regard, it has been shown that both the local or intraperitoneal injection of anti–TGF–β in mice with arthritis increases disease severity and proinflammatory cytokine levels.32,33
Role of TGF–β in Animal Models of Arthritis.
| Animal modelAnti-inflammatory effect | Animal modelProinflammatory effect | Administration | Ref. |
| Mice with CIARats with SCW | TGF–β 1 IP | 26–28 | |
| Mice with CIA | Mesenchymal cells, TGF–β IP | 30 | |
| Mice with CIA | Anti-TGF–β local and IP | 32,33 | |
| Healthy mice and rats | TGF–β 1 local | 34–36 | |
| Rats with SCW | Anti-TGF–β local | 37 | |
| Mice with CAIA | Anti-TGF–β RI | 2 |
Ab, antibodies; CAIA, arthritis induced with anti-collagen antibodies; CIA, collagen-induced arthritis; TGF–β 1, transforming growth factor-beta 1; IP, intraperitoneal; SCW, arthritis induced by Streptococcus.
However, there are also many studies that exhibit a completely opposite role of TGF–β. In fact, repeated injections of TGF–β 1 in the healthy mice induced joint inflammation and synovial hyperplasia.34 Similarly, joint injection of TGF–β 1 in rats induces neutrophil recruitment, erythema, synovial proliferation and infiltration, mainly by T lymphocytes.35,36 Several studies support this concept of TGF–β as a proinflammatory agent. Treatment with anti–TGF–β antibodies directly into the joint of rats with arthritis induced through a SCW model inhibits synovial inflammation, bone resorption and the production of proinflammatory cytokines.37 Other authors have shown that treatment with anti-TGF-β RI-antibodies induced arthritis in mice with anti-collagen Ab prevents arthritis, the development of hyperplasia, synovial inflammation and angiogenesis.2 Among the therapeutic strategies described in the literature to block TGF–β specifically, p17 is of note.38 A study recently published by our group demonstrated that selective blockade of TGF–β with p17 moderately slows the signs of arthritis in the mouse model of CIA. However, this blockade has no effect on the differentiation and functionality of different populations of T cells, cytokines or the degree of osteocartilaginous destruction and concluded that TGF–β does not have a role in the CIA mouse model.39
The role of TGF–β differentiation of murine T cells has been extensively studied. In mice, it has been demonstrated that the specific overexpression of TGF–β in T cells leads to the generation of T cells with regulatory function and protects mice deficient in IL-2 for the development of severe systemic inflammation and autoimmunity.40,41 In the CIA model, Treg cell transfer at the time of immunization reduces the severity of arthritis.42,43 However, the role of TGF–β in the differentiation of Th17 cells is not completely defined. Initially, it was reported that TGF–β and IL-6, through the induction of IL-21, were able to promote T α β CD4+ naïve cell differentiation into Th17 cells via activation of the transducer factor signal STAT-3 and transcription factors Ror-gamma t and Ror α γ.44,45 Other studies have shown that IL-21, a member of the family of IL-2 cytokines, cooperates with TGF–β to induce differentiation of Th17 cells even in the absence of IL-6,46,47 while IL-23, produced by activated dendritic cells induces the survival and expansion of previously differentiated Th17 cells.48 However, a recent study demonstrated that the differentiation of Th17 cells in a murine model of experimental allergic encephalitis (EAE) can occur in the absence of TGF–β and in the presence of IL-6, IL-23 and IL-1 β,49 similar to that originally proposed in humans. These Th17 cells induced by IL-23 coexpress with a pathogenic phenotype γ Ror-gamma t and T-bet and can result in γ IL-17/IFN-producing cells, unlike Th17 cells induced by TGF–β, considered as non-pathogenic. Other authors have suggested that TGF–β does not act directly to promote differentiation of murine Th17 cells, but acts indirectly to regulate IL-17 by removal of the factors that contribute to the differentiation of Th1 and Th2 cells.50 Very recently it has been reported that Th17 cells developed by a mechanism dependent on IL-23, IL-6 and TGF–β 1 are pathogenic and produce large amounts of TGF–β 3.51 In addition, TGF–β 3 Th17 cells have a pathogenic role in EAE models of colitis, similar to the pathogenicity of Th17 differentiated in the presence of IL-6 and TGF–β 1 and produce higher amounts of IL-23. Moreover, CD8 + T cells in the rheumatoid synovium induce the synthesis of IFN–γ and IL-17, and have demonstrated their role in a chronic synovitis model K/BxN mouse.52 TGF-β acts on CD8+ cells, inducing the production of Foxp3 and IL-17, resulting in CD8+ Treg and CD8+ IL-17+, respectively.53,54 However, its role in triggering CIA55,56 shows mixed results, indicating that these cells may not be involved in the onset of disease.
The role of TGF–β in animal models of RA, therefore, is not well defined and the discrepancy in the results for effect on the different cell types in the rheumatoid synovium indicate an impairment of the factors responsible for the Smad protein dependent intracellular pathway. These changes result in abnormal responses of TGF–β, conditioning altered cell behavior. Smad-3−/− animals exhibit an exaggerated inflammatory response, while the deficit of Smad-2−/− is lethal due to defects in embryonic development.57 Also, transgenic mice for Smad-7 are associated with increased production of cytokines by T helper (Th) 1 and Th2 cells. Some authors have reported that Smad-2 is significantly reduced in the cartilage during the progression of osteoarthritis (OA) in several animal models,58 while other studies show an increase in the expression of TGF-RII β and synovial fibroblasts of RA patients compared with OA patients.59
In humans, almost all the cellular components of the rheumatoid synovium may secrete TGF–β and besides exerting action on mesenchymal cells, this cytokine plays a modulatory effect on inflammatory cells. In in vitro studies with cells of the rheumatoid synovium TGF–β dependent effects are disparate, behaving as immunoregulatory inducer or inhibitor factor of the inflammatory response according to the surrounding conditions. Among the anti-inflammatory effects, the inhibition of lymphocyte proliferation and superoxide production by macrophages are included.60 However, it can also act as a proinflammatory factor in stimulating the secretion of cytokines such as TNF–α and IL-1, acting as a potent chemoattractant for neutrophils, activating the expression of chemokines, MMP and,61,62 inducing expression of VEGF,63 essential in the development of angiogenesis in RA, and modulating apoptosis of SF in a poorly defined mechanism. In this regard, TGF–β inhibits Fas expression and increases Bcl-2 and proto-Bclx, although other authors suggested PI3K and Akt activation as responsible for the antiapoptotic effect of TGF–β.64
The mononuclear cells from rheumatoid synovial tissue in culture produce β TGF-1 and its presence has been demonstrated in the synovial tissue and fluid.65 Microarray studies suggest an increase in the TGF-signaling pathway in SF-β of RA patients as compared to patients with OA, in particular TGF-1 and its receptor β (TGF–β RI), accompanied by an increased expression of MMP. This increased TGF–β 1 expression correlates directly with clinical markers of disease activity such as C-reactive protein serum activity and ACR66 criteria, indicating a direct correlation between TGF-1 and inflammation β. In this sense, the described activation of TGF–β signaling pathways in mononuclear cells in rheumatoid inflammatory aggregates indicates a possible pathogenetic involvement of this cytokine in the development of the disease.39
The SM in RA contains lots of T67 cells. However, it has been difficult to define the role these play in the maintenance and propagation of joint inflammation. The use in the treatment of RA of a fusion protein of human CTLA-4 inhibiting T cell costimulation via CD28 has provided an indirect but convincing evidence for the importance of T cells in RA.68 The role of TGF–β in differentiating different populations of human T cells is manifold. On one hand, it has been shown that signaling through TGF–β 1 protects regulatory T (Treg) cells from apoptosis69 and is absolutely necessary to induce the expression of Foxp3, as it has been shown that impaired CD4+ cells due to lack of TGF–β 1 can not generate Treg cells in vivo and in vitro.70–73 Treg cells can develop from thymic CD4+ T cell precursors in the presence of TGF–β and IL-2, resulting in natural Treg cells. In the periphery, the naïve CD4+ cells can become inducible Treg cells via STAT signaling-5 in the presence of TGF–β, increasing Foxp3 expression (Fig. 1). The Foxp3 promoter regulation depends on, among other things, on the NF-AT transcription factor and Smad-3 signaling induced by TGF–β.74 In fact, TGF–β mediates Foxp3 expression by directly binding to the promoter of Smad. Treg cells secrete low amounts of IL-2 and IFN–γ, while producing high amounts of IL-10, IL-35 and TGF–β, essential to regulate the suppression of T cells.75 Recently, it has been described that, in the absence of TGF–β1, functional development of Treg in cancer cells could be produced by a compensatory mechanism dependent on the expression of TGF-β 2 and TGF–β 3 in the thymus and periphery.76,77 However, it is unknown whether this mechanism occurs in RA.
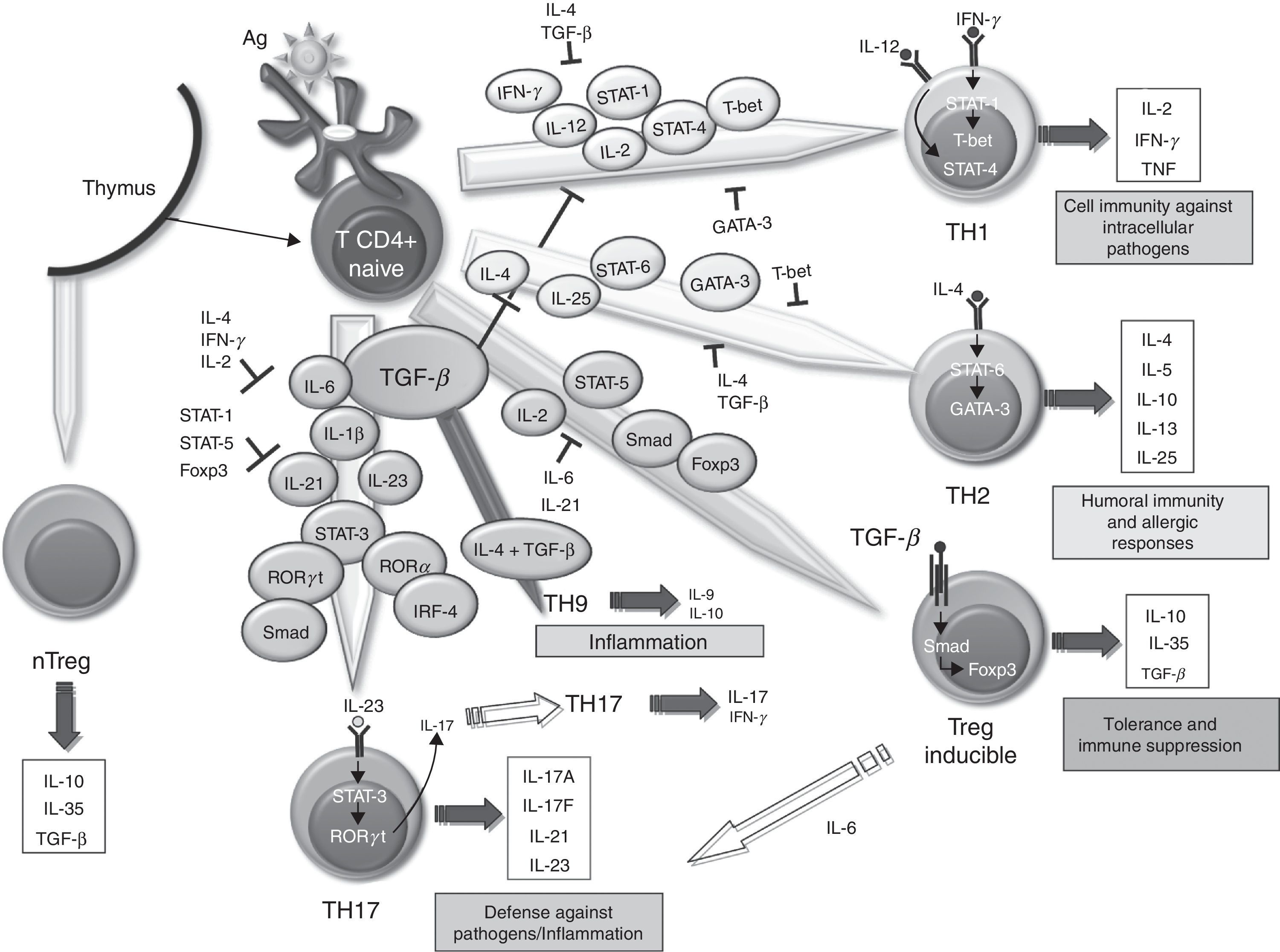
Role of TGF–β in the differentiation of effector T cells. Effect of TGF–β and molecules involved in the differentiation and regulation of the different populations of effector T cells in relation to their role in the immune system. IL, interleukin; TGF–β, transforming growth factor-beta; Treg, regulatory T cells.
Classically, it was assumed that RA was a disease mediated by Th1 cells producing IFN–γ in the absence of Th2 cytokines. In the differentiation of both cell types, TGF–β acts as a negative regulator by inhibiting T-bet and GATA-3 transcription factors,78,79 respectively. However, over the past 10 years, it has become increasingly important as Th17 cells promote the inflammatory response in RA. In human Th17 differentiation, several mechanisms may be involved. First, it was determined that the differentiation of human Th17 cells occurred in the absence of TGF–β80 and that the combination of IL-1 β, IL-6 and IL-23 was able to act on memory T cells to produce IL-17. However, other studies disclose a role for TGF–β, which together with IL-21 and expression of Rorc2 promote differentiation of naïve CD4+ to Th17 cells.81 In this regard, TGF–β through IL-21 suppresses the induction of T-bet. Recent studies point to an interaction of mesenchymal stem cells derived from bone marrow or synovial fibroblasts with T cells as promoting activation and expansion of Th17 cells, which may contribute to the chronicity of RA.82 Recently, we have described a new subtype of Th cells, Th9, producing IL-9 and IL-10. The differentiation of these cells from naïve T cells is TGF–β and IL-4 dependent,83 although the treatment of Th2 cells with TGF–β is also capable of producing Th9 cells. In spite of the production of IL-10, when not expressing Foxp3, they are not considered regulatory cells, but inflammation-promoting cells. In fact, although its role in RA has not been investigated in other models of inflammatory disease, such as EAE, it has been noted that they can act as inducers of inflammation.84 It has also been shown that the stimulation with TGF–β and IL-1-induced CD4 β IL-9+/+ IL-17 in patients with autoimmune diabetes,85 hence it has a possible role in other autoimmune diseases.
ConclusionTGF-β is a cytokine involved in numerous biological processes. Of the 3 isoforms of TGF–β that exist in mammals, TGF–β 1 plays the most important role in the immune system, particularly in the control of proliferation, activation and differentiation of T cells; one study to try to define the role of TGF–β in RA have shown mixed findings. The differences of these findings could be due to the animal model used, the time since onset of the disease in the study and the protocol used for inhibition of the cytokine.
Ethical ResponsibilitiesProtection of people and animalsThe authors declare that this research has not performed experiments on humans or animals.
Data privacyThe authors state that patient data does not appear in this article.
Right to privacy and informed consentThe authors state that no patient data appears in this article.
FinancingThis project was funded by a grant from the Spanish Foundation of Rheumatology (FER 2009) in the form of aid for research projects not funded by public agencies. Elena Gonzalo Gil has received funding from a grant from the Spanish Foundation of Rheumatology (FER) in the form of aid to supplement research projects already funded.
Conflicts of InterestThe authors have no conflicts of interest.
The authors thank Drs. José Luis Pablos Alvarez and Gabriel Criado Carrasco, for their assistance in the design of experiments and critical review of the results.
Please cite this article as: Gonzalo-Gil E, Galindo-Izquierdo M. Papel del factor de crecimiento transformador-beta (TGF-β) en la fisiopatología de la artritis reumatoide. Reumatol Clin. 2014;10:174–179.


