



Mucolypidosis (ML) are a group of metabolic diseases with autosomal recessive inheritance belonging to the group of lysosomes storage diseases. ML has classically been classified into 4 types (I–IV), but the ICD-10 considers the type IA a glucoproteinosis and ML IVA a gangliosidosis, so ML strictly would be type II and III. In those conditions, the alteration is in the lysosomal enzyme N-acetylglucosamine-1-phosphotransferase, reduced in ML II and absent or altered in MP IIIA and IIIC1 respectively. We present the case of a male affected with MP III or pseudo-Hurler polydystrophia, a rare disease with characteristic bone radiographic abnormalities.
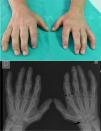
Clinical CaseThe patient is a 47-year-old male, with healthy parents, non consanguineous, with a 42-year-old sister with height stunting and spinal surgery (unspecified), a 40-year-old brother with arthroplasty of both hips and a sister, 37, healthy. At 3 years of age, he presented difficulty bending his fingers, so he was referred for study to the University Hospital of La Paz, where he was diagnosed with ML type III and followed up to 14 years. At this age, he had lost two school years, finishing only primary education. He underwent a bilateral hip arthroplasty (right 2008, left 2011). No cardiac symptoms. No referred mechanical low back pain in the past 2 years, yielding to common analgesics, which he takes on demand. On physical examination: height 154cm, weight 57.2kg. Coarse facial features, claw hands (Fig. 1), limited in maximum flexion and extension of the fingers, without skin thickening. Asymmetry of lumbar folds with elevation of the left hemipelvis. No tenderness of lumbar spinous processes, exploring the sensitivity and mobility is normal and root stretching maneuvers are negative.
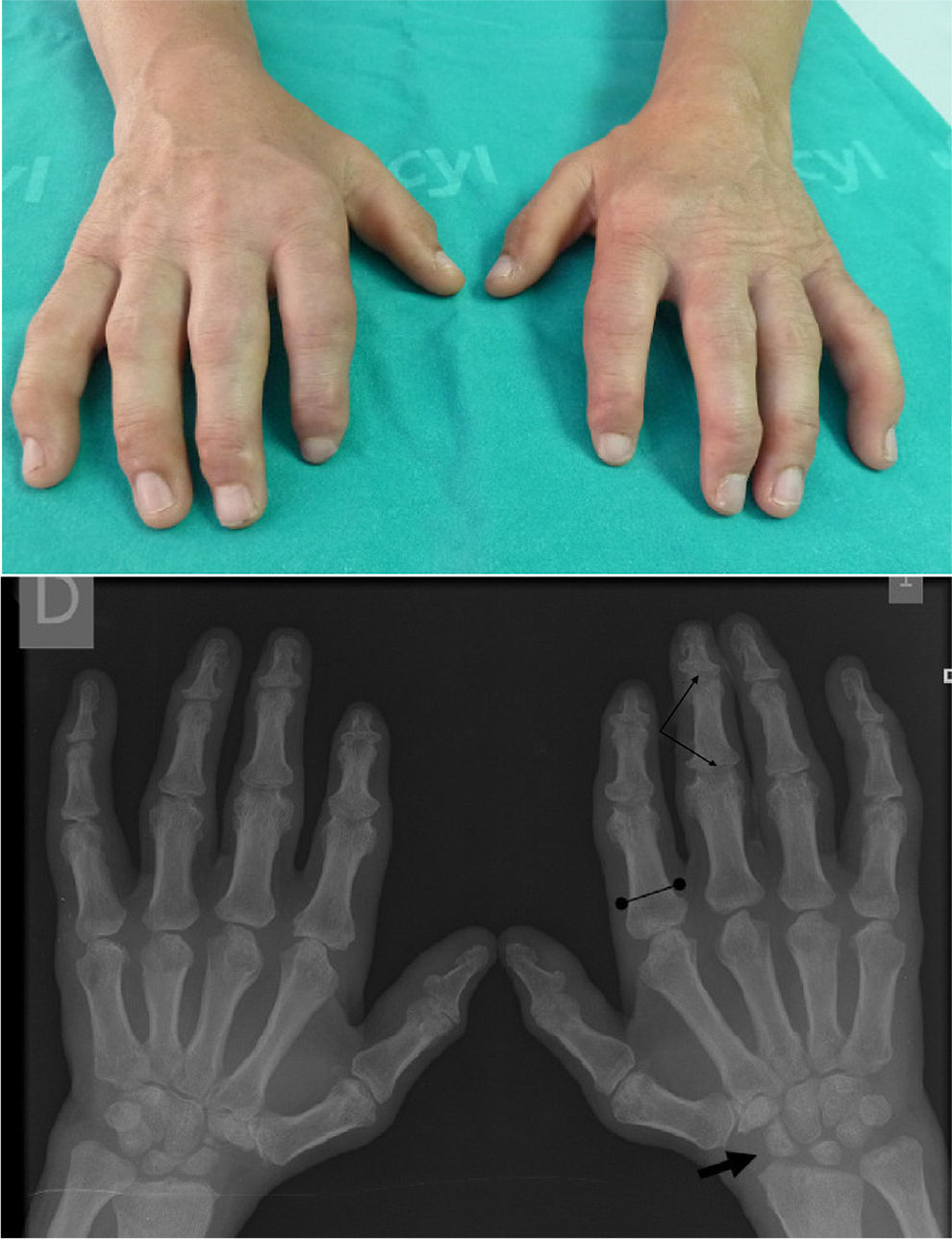
Claw Hand: impossible flexion and maximum extension, without objetive skin thickening or joint involvement. Hand X ray: small and irregular carpus bones (arrow) and wide proximal phalanges (bounded line). The attitude of the claw fingers produces an image of pseudoimpingement in the PIP and DIP joints (thin arrows).
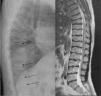
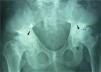
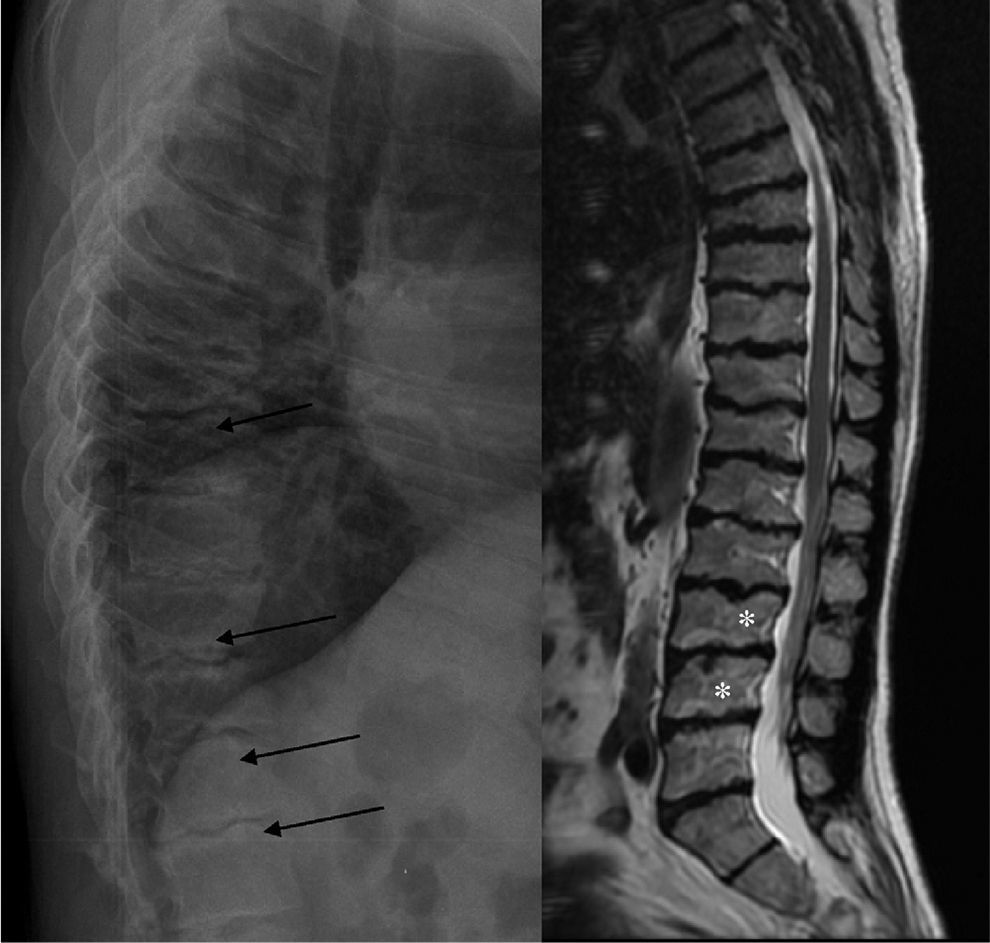
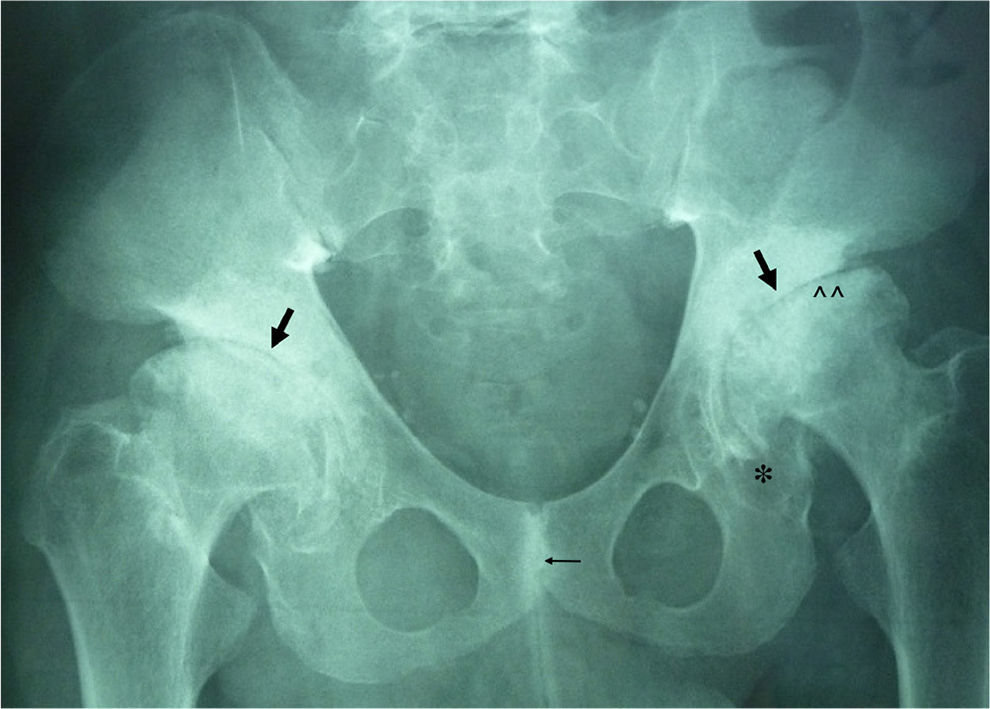
ML III occurs approximately between 3 and 5 years of age, with skeletal and facial abnormalities, short stature, normal intelligence or mild mental retardation, corneal opacity and scoliosis, unlike other forms of ML that have visceral involvement and a gloomy vital prognosis in childhood.2,3 In ML III, evolution is slow, and patients can reach up to the fifth decade of life.4 Bone alterations in children may be confused with juvenile idiopathic arthritis or scleroderma, primarily by involvement of the hands.5,6 The characteristic radiologic findings of the hands are small and irregular carpal bones and relatively wide proximal phalanges5 (Fig. 1). In the lumbar spine, vertebral dysplasia with irregular delineation of the vertebral bodies3 is confirmed on MRI (Fig. 2). In the pelvis, progressive hip dysplasia, with a flattened acetabulum and femoral head destruction with secondary coxarthrosis is seen2 (Fig. 3).
Genetic counseling and symptomatic treatment are available as therapeutic tools. In some cases of ML, pamidronate has been used intravenously in order to reduce3,7 osteodystrophy.
Ethical ResponsibilitiesProtection of people and animalsThe authors declare that experiments were not performed on humans or animals.
Data confidentialityThe authors declare that they have followed the protocols of their workplace regarding the publication of data from patients, and all patients included in the study have received sufficient information and gave written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained informed consent from patients and/or subjects referred to in the article. This document is in the possession of the corresponding author.
Conflict of InterestThe authors have no disclosures to make.
Please cite this article as: Pantoja Zarza L, Diez Morrondo C. Alteraciones óseas en la mucolipidosis III. Reumatol Clin. 2014;10:340–341.


