

Systemic sclerosis sine scleroderma (ssSSc) is a form of systemic sclerosis that is characterized by Raynaud's phenomenon (RP), visceral involvement without thickening of skin and anticentromere antibodies (ACA). We studied 10 ssSsc patients with a prevalence of 2%. The clinical signs were: RP 9/10, esophageal manifestations 8/10, pulmonary arterial hypertension 4/10, interstitial lung disease 4/10, cardiac signs 3/10 and ACA 8/10.
ConclusionIn patients with RP, esophageal dysmotility, interstitial lung disease and pulmonary arterial hypertension should be tested for ACA in order to establish a prompt diagnosis and treatment of ssSSc.
La esclerosis sistémica sine esclerodermia (ESse) es una forma de esclerosis sistémica caracterizada por fenómeno de Raynaud (FR), afección visceral sin endurecimiento de la piel y anticuerpos anti-centrómeros (AAC). Se estudiaron a 10 pacientes con ESse, con prevalencia del 2%. Manifestaciones clínicas: FR 9/10, esofágica 8/10, hipertensión arterial pulmonar 4/10, neumopatía intersticial 4/10, cardiaca 3/10 y AAC 8/10.
ConclusiónEn pacientes con FR, dismotilidad esofágica, neumopatía intersticial e hipertensión arterial pulmonar se debe investigar AAC y establecer un diagnóstico y tratamiento oportuno de ESse.
Systemic sclerosis (SSc) is characterized by the excessive production of collagen in the skin and internal organs, including lung, heart, esophagus and gastrointestinal tract, among others, as well as vascular changes, such as pulmonary arterial hypertension (PAH), renal crisis and Raynaud's phenomenon (RP), in addition to immune disorders.1,2 Depending on the skin involvement, SSc is defined as diffuse SSc in patients with hardening of the skin proximal to elbows and knees, aside from the trunk, and limited SSc (lSSc) in patients with hardening of the skin distal from elbows and knees, as well as on the face.2,3 Systemic sclerosis is classified as prescleroderma, lSSc, the diffuse form and SSc sine scleroderma (ssSSc). The latter is distinguished as it affects organs, but without hardening of the skin; its frequency is 2%–8% and it depends on the organ involved: esophagus (53%–86%), lung (25%–57%) and kidneys (2.5%–3.7%).1
Clinical ObservationWe did a retrospective study of 10 patients with ssSSc from a cohort of 500 SSc patients treated during the period from 2005 to 2015. The inclusion criteria called for patients without skin sclerosis, but with the following manifestations: (1) RP or equivalents (pitting or ulcers in the fleshy part of the fingertip or capillaroscopic changes); (2) positive test for antinuclear antibodies (ANA); and (3) at least 1 of the following visceral manifestations, which included hypomotility of the distal esophagus and small intestine, interstitial lung disease (ILD), PAH, cardiac involvement typical of scleroderma or scleroderma renal crisis, but no connective tissue disease or other disease that could explain the abovementioned manifestations.4 We excluded patients with other forms of SSc. All of the patients underwent upper gastrointestinal endoscopy, esophageal manometry, upper gastrointestinal series, intestinal transit, high-resolution pulmonary computed tomography, respiratory function tests, Doppler echocardiography, right cardiac catheterization (performed in 4 patients), electrocardiography and Holter monitoring (the latter in 4 patients), as well as detection of antinuclear antibodies (ANA) by immunofluorescence (HEp-2 cells as substrates) and antibodies to extractable nuclear antigens: anti-topoisomerase, anti-Ro/La, anti-ribonucleoprotein (RNP), anti-Smith (Sm), anti-Jo-1 and anticentromere antibodies (ACA). Esophageal biopsy was carried out in all the patients and lung biopsy in one. Capillaroscopy was not performed.
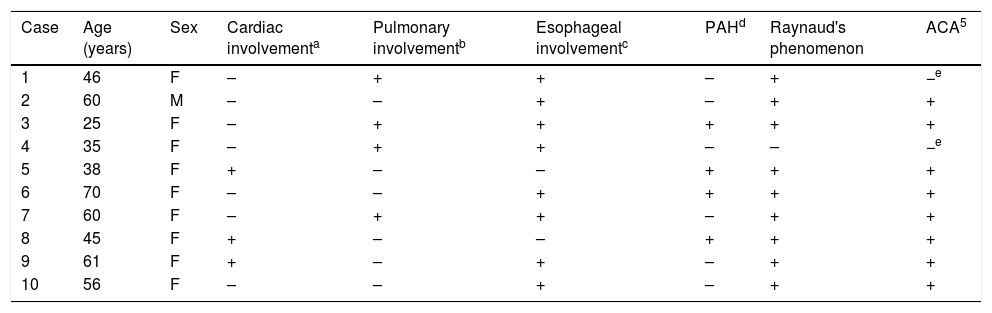
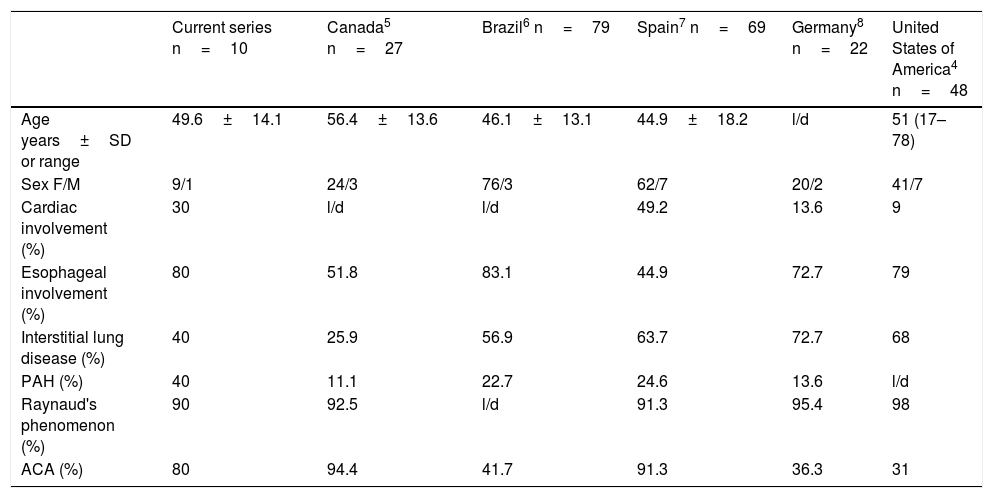
ResultsTable 1 shows the clinical characteristics and autoantibodies detected in these patients. Nine of the 10 patients were women. The average age was 49.6±14.1 years, the mean duration of the disease 8.8±5.4 years and the delay in diagnosis was 2.6±1.5 years. The main clinical manifestations observed were: RP (9/10); esophageal involvement (8/10) with moderate to severe gastroesophageal reflux; pulmonary involvement (4/10) with ILD and/or PAH (4/10; moderate in 4 patients and mild in 1); cardiac involvement (3/10) with left ventricular diastolic dysfunction—grade II in 2 patients and III in 1 patient, conduction disturbances and heart failure (1/10). The ANA titers ranged between 1:320 and 1:1280 (3 patients had a nucleolar pattern, especially patients 1 and 4 of Table 1). Patient 1 was also positive for anti-Scl-70. Most of the patients (9/10) were positive for ACA. Table 2 shows the main clinical manifestations of ssSSc and compares them with those described in other reported series.
Clinical Characteristics and Autoantibodies in Patients With Systemic Sclerosis Sine Scleroderma.
| Case | Age (years) | Sex | Cardiac involvementa | Pulmonary involvementb | Esophageal involvementc | PAHd | Raynaud's phenomenon | ACA5 |
|---|---|---|---|---|---|---|---|---|
| 1 | 46 | F | – | + | + | – | + | −e |
| 2 | 60 | M | – | – | + | – | + | + |
| 3 | 25 | F | – | + | + | + | + | + |
| 4 | 35 | F | – | + | + | – | – | −e |
| 5 | 38 | F | + | – | – | + | + | + |
| 6 | 70 | F | – | – | + | + | + | + |
| 7 | 60 | F | – | + | + | – | + | + |
| 8 | 45 | F | + | – | – | + | + | + |
| 9 | 61 | F | + | – | + | – | + | + |
| 10 | 56 | F | – | – | + | – | + | + |
ACA, anticentromere antibodies; F, female; M, male; PAH, pulmonary arterial hypertension.
Left ventricular diastolic dysfunction, heart failure and valve diseases (tricuspid and pulmonary insufficiency) evaluated by Doppler echocardiography.
Bilateral pulmonary fibrosis affecting the lung bases, detected by high-resolution computed tomography and respiratory function tests.
Systemic Involvement in Systemic Sclerosis Sine Scleroderma and Comparison With Other Series.
| Current series n=10 | Canada5 n=27 | Brazil6 n=79 | Spain7 n=69 | Germany8 n=22 | United States of America4 n=48 | |
|---|---|---|---|---|---|---|
| Age years±SD or range | 49.6±14.1 | 56.4±13.6 | 46.1±13.1 | 44.9±18.2 | l/d | 51 (17–78) |
| Sex F/M | 9/1 | 24/3 | 76/3 | 62/7 | 20/2 | 41/7 |
| Cardiac involvement (%) | 30 | l/d | l/d | 49.2 | 13.6 | 9 |
| Esophageal involvement (%) | 80 | 51.8 | 83.1 | 44.9 | 72.7 | 79 |
| Interstitial lung disease (%) | 40 | 25.9 | 56.9 | 63.7 | 72.7 | 68 |
| PAH (%) | 40 | 11.1 | 22.7 | 24.6 | 13.6 | l/d |
| Raynaud's phenomenon (%) | 90 | 92.5 | l/d | 91.3 | 95.4 | 98 |
| ACA (%) | 80 | 94.4 | 41.7 | 91.3 | 36.3 | 31 |
ACA, anticentromere antibodies; F, female; l/d, lack of data; M, male; PAH, pulmonary arterial hypertension; SD, standard deviation.
Systemic sclerosis sine scleroderma (ssSSc) is an uncommon form of SSc. In our study, the prevalence of ssSSc was 2%, ranging from 1.5% to 10%, similar to data reported previously.1,4–8 This prevalence is probably underestimated due to the absence of skin involvement in these patients, a fact that means that there is often a delay in the diagnosis. In our patients, the average time to the diagnosis of ssSSc was 2.6±1.5 years. Findings such as pulmonary involvement (ILD and/or PAH) and RP, observed in some of our patients, led to the suspicion of the diagnosis of ssSSc. In the current classification of SSc,7 ssSSc is considered a subtype of SSc; however, some authors are still not sure whether it is a different form of SSc or part of the clinical spectrum of lSSc.4–6 There are clinical similarities between ssSSc and lSSc, except for the presence of telangiectasia observed in the former.6 Raynaud's phenomenon was the clinical manifestation most frequently found in our patients (90%). The organ most commonly affected was the esophagus, as a result of gastroesophageal reflux, followed by the lung, with ILD and PAH, which were detected in 40%, respectively, and the heart (30%) with manifestations of left ventricular diastolic dysfunction and conduction disturbances. These clinical signs are similar to those reported in other series of patients, with the exception of renal crisis, which in our series was not observed in any case.4
Anticentromere antibodies were identified in 80%, as seen in other patient populations, and the remainder of the patients had ANA.6 In ssSSc, as the skin is not affected, in general, the diagnosis and treatment are delayed, a fact that can lead to higher rates of morbidity and mortality.9 The treatment of ssSSc is similar to that of patients with SSc, with emphasis on the organ involved.5,8 It is considered a mild form of SSc with a good prognosis,6 although that will depend on the affected organ. For some authors the prognosis of ssSSc is similar to that of lSSc.5,8,9
In short, patients with ILD or PAH and esophageal dysmotility should be tested for ACA and ssSSc must be taken into account in order to establish a prompt diagnosis and treatment.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of InterestsThe authors declare that they have no conflict of interests.
Please cite this article as: Vera-Lastra O, Sauceda-Casas CA, Cruz Domínguez MP, Mendoza Alvarez SA, Sepulceda-Delgado J. Esclerosis sistémica sin esclerodermia en pacientes mexicanos. Serie de casos. Reumatol Clin. 2018;14:230–232.


