





La psoriasis y la artritis psoriásica son enfermedades autoinmunitarias que comparten mecanismos patogénicos. No se conoce la causa ni tampoco se han desvelado completamente las bases moleculares y celulares implicadas en la patogenia de estas enfermedades. Sin embargo, numerosas evidencias sugieren que la generación de linfocitos T CD8 citotóxicos autorreactivos y la activación de las respuestas inmunitarias mediadas por linfocitos Th1 y Th17 participan en el desencadenamiento y perpetuación de la artritis psoriásica. Las citocinas que se producen durante la respuesta Th1 y Th17, como el TNF-α o la IL-17, desempañan un papel destacado en la respuesta inflamatoria crónica que se observa en las articulaciones de los individuos afectados. Es previsible que el completo esclarecimiento de la patogenia de esta enfermedad nos permita el desarrollo de nuevos tratamientos y una mejora del pronóstico de los pacientes.
Psoriasis and psoriatic arthritis are autoimmune diseases that share common pathogenic mechanisms. The cause and the pathogenesis of these diseases are unknown; however, there is increasing evidence which suggest that cytotoxic CD8T cells, Th1 and Th17 may be involved in the pathogenesis of these diseases. Cytokines produced as the result of Th1 and Th17 responses, such as TNF-α and IL-17, play a key role in the chronic inflammatory response observed in the joints of psoriatic arthritis patients. It is conceivable that unraveling of the pathogenesis of this disease may lead to the development of new therapeutic approaches and may improve the prognosis of patients.
La artritis psoriásica es una enfermedad inflamatoria crónica que presenta una prevalencia elevada, que afecta a un 5-30% de los individuos con psoriasis. Fue inicialmente fue definida en 1973 por Moll y Wright como una artritis inflamatoria periférica y/o espondilitis asociada con psoriasis y serología negativa para el factor reumatoide1. Desde entonces se ha descrito un amplio espectro de presentaciones clínicas, lo que ha derivado en el desarrollo de criterios que permiten distinguirla de otras enfermedades reumáticas2.
No se ha desvelado completamente la patogenia de estas enfermedades, pero todas las evidencias indican que la psoriasis y la artritis psoriásica son enfermedades autoinmunitarias que comparten mecanismos patogénicos comunes. En las enfermedades autoinmunitarias se produce una respuesta inmunitaria adaptativa mediada por linfocitos contra algún antígeno (Ag) propio o auto-Ag. Aunque existe casi un centenar de enfermedades autoinmunitarias, algunas bastante prevalentes, no se conocen las causas ni en gran parte los mecanismos implicados en el desarrollo de estas enfermedades. Esto hace que todavía se genere mucho debate sobre la naturaleza misma de estas enfermedades.
Respuesta inmunitaria y autoinmunidadEl sistema inmunitario está formado por dos componentes que trabajan coordinadamente, la inmunidad innata y la inmunidad adaptativa3. Las células de la inmunidad innata, por ejemplo los macrófagos, expresan receptores para moléculas conservadas que les permite reconocer grupos o familias enteras de patógenos. Estos receptores han sido generados en el curso de la evolución para reconocer los Ag extraños y no los propios. La ausencia de receptores para nuestros propios Ag es la responsable de que la inmunidad innata no sea la causante del desencadenamiento de las enfermedades autoinmunitarias. La respuesta inmunitaria adaptativa está mediada por los linfocitos, los cuales pueden reconocer cualquier Ag extraño. Para ello, los linfocitos B y T generan por azar en cada individuo un número prácticamente ilimitado de receptores para el Ag. Dado que se produce por azar, es inevitable que se generen linfocitos autorreactivos capaces de reconocer nuestros propios Ag. Estos linfocitos autorreactivos deber ser eliminados o inactivados para asegurar la tolerancia a nuestras propias moléculas. Las enfermedades autoinmunitarias se producen precisamente porque en algunos individuos fracasa este mecanismo de tolerancia contra los auto-Ag. Por tanto, se piensa que la inmunidad adaptativa, y no la innata, es la causante del desencadenamiento de las enfermedades autoinmunitarias, como la artritis psoriásica. La inmunidad innata puede participar en la patogenia de la enfermedad, pero siempre lo hace invocada por los linfocitos autorreactivos. Esta idea, aunque está ampliamente aceptada, no es compartida por todos. En la artritis psoriásica en particular se ha cuestionado la naturaleza autoinmunitaria de la enfermedad y se ha postulado un mayor peso de la inmunidad innata en su desarrollo. De ahí ha surgido el concepto del complejo enteso-sinovial, aunque este concepto solo es capaz de explicar una parte de las características de la enfermedad4.
Existen diferentes respuestas inmunitarias adaptativas implicadas en la patogenia de las enfermedades autoinmunitariasLa inmunidad adaptativa desarrolla diferentes tipos de respuestas debido a que existen diferentes tipos de patógenos. Para el sistema inmunitario no es esencialmente importante la naturaleza biológica de los patógenos, sino si es intracelular o extracelular, y si es intracelular, si está presente en el citoplasma o en el lisosoma de nuestras células. Según donde se encuentre, producirá una respuesta adaptativa diferente (fig. 1). Los patógenos intracelulares citoplasmáticos, como los virus, son reconocidos porque los péptidos derivados de las proteínas citoplasmáticas son presentados por las moléculas de MHC de clase I (p. ej., HLA-Cw*0602) a los linfocitos T CD8 o citotóxicos. Estos linfocitos se denominan así porque cuando reconocen un péptido extraño a través de su receptor (TCR) matan las células infectadas. Los patógenos intracelulares que viven en los lisosomas de los macrófagos (como los Micobacterium) son reconocidos porque sus Ag son presentadas por las moléculas MHC de clase II a un tipo de linfocitos T CD4 denominado Th1. Estos linfocitos activan a los macrófagos infectados para que eliminen al microorganismo. En esta respuesta, la activación de los macrófagos por los linfocitos Th1 causa la liberación de TNF-α, lo que genera una respuesta inflamatoria intensa. Los patógenos extracelulares son reconocidos por un tipo linfocitos T CD4 denominados Th2, los cuales colaboran con los linfocitos B en la producción de anticuerpos (Ac) contra estos microorganismos. Se ha descrito recientemente otro tipo de linfocitos T CD4, denominado Th17, que participa en las respuestas contra ciertos patógenos extracelulares favoreciendo la respuesta mediada por neutrófilos, aunque las bases celulares de esta respuesta no son aún bien conocidas.
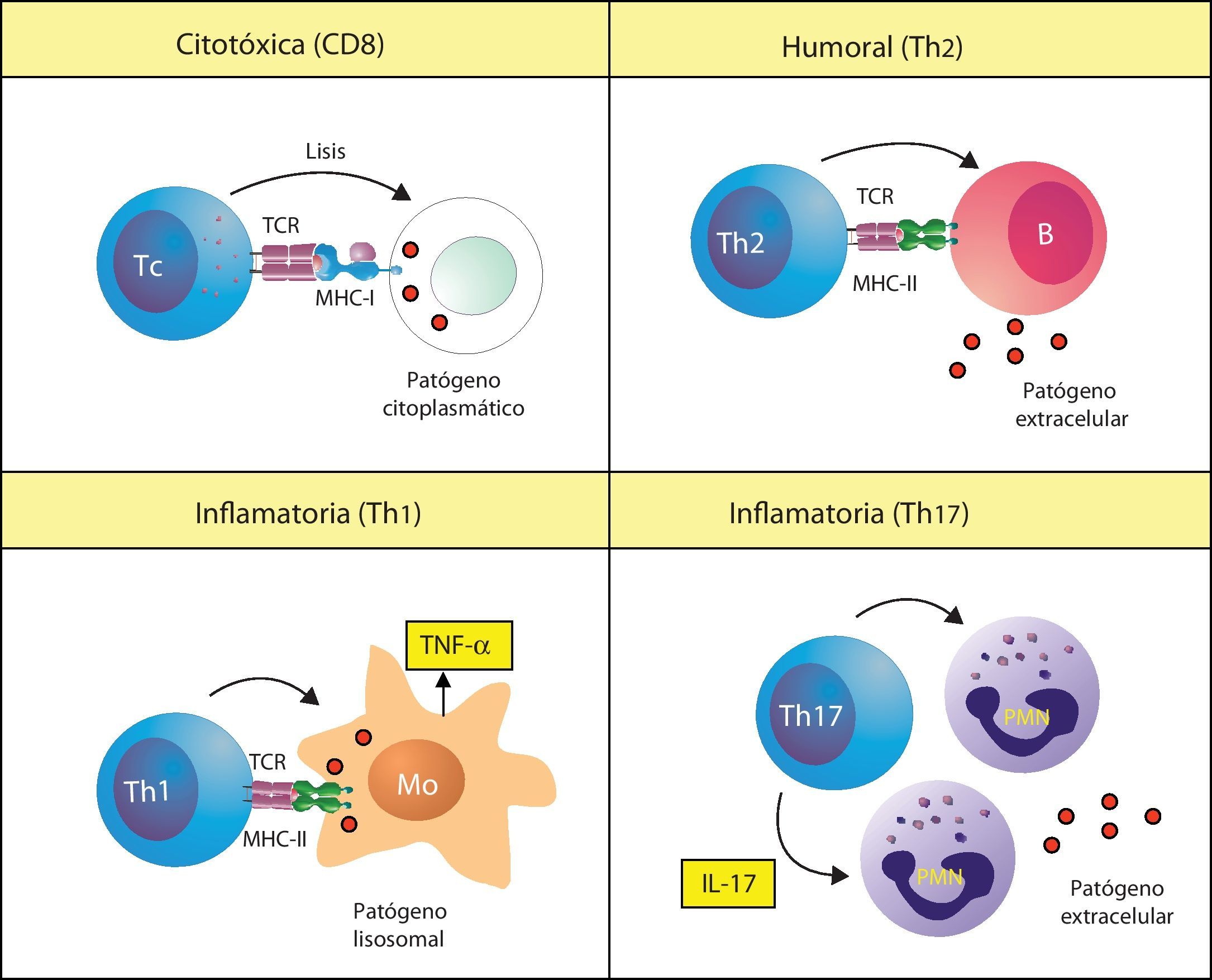
Tipos de respuestas inmunitarias adaptativas. El sistema inmunitario produce respuestas inmunitarias adaptativas diferentes, denominadas citotóxica, Th1, Th2 o Th17, cada una de las cuales es específica para un tipo de patógeno. Estas mismas respuestas también se desencadenan en la autoinmunidad. MHC-I: MHC de clase I; MHC-II: MHC de clase II; Mo: macrófago; PMN: célula polimorfonuclear o neutrófilo; Tc: linfocito T citotóxico; TCR: receptor de los linfocitos T.
En las enfermedades autoinmunitarias, se reconoce como extraño algo propio, pero los mecanismos inmunitarios adaptativos que se generan para eliminar ese Ag son iguales que contra los patógenos. Por ejemplo, si el auto-Ag se encuentra en el citoplasma, como en el caso de la diabetes mellitus insulinodependiente, se producirá una respuesta citotóxica mediada por linfocitos T CD8 que destruyen las células β del páncreas. Si el auto-Ag es extracelular, la respuesta será tipo Th2 e implicará la producción de auto-Ac, como ocurre, por ejemplo, en el lupus eritematoso sistémico. Si el auto-Ag es fagocitado por los macrófagos, este estará presente en sus lisosomas, por lo que se inducirá una respuesta Th1, como se piensa que ocurre en la artritis reumatoide. Una de las consecuencias de la activación de los macrófagos es la producción crónica de TNF-α, que causa una inflamación crónica de los tejidos afectados. Recientemente, se ha descubierto que las células Th17 también pueden causar inflamación y autoinmunidad, y que pueden participar en la patogenia de enfermedades autoinmunitarias inflamatorias, como la artritis reumatoide5. Sin embargo, existe una característica fundamental que diferencia las respuestas autoinmunitarias. En estas, el auto-Ag generalmente persiste, ya que es sintetizado constantemente en nuestras células, por lo que el sistema inmunitario es generalmente incapaz de eliminarlo. Por ello las enfermedades autoinmunitarias suelen ser crónicas y duran toda la vida.
Etiología de la artritis psoriásicaNo se conoce la causa de las enfermedades autoinmunitarias; sin embargo, se piensa que se producen por causas ambientales que actúan sobre individuos genéticamente predispuestos. Tampoco se conoce el componente ambiental causante del desencadenamiento de prácticamente ninguna enfermedad autoinmunitaria, pero se ha postulado que, en la mayoría de los casos, el responsable puede ser un microorganismo, ya que se conocen diversos mecanismos por los que un microbio puede romper la tolerancia a nuestros propios Ag. La fiebre reumática es la única enfermedad autoinmunitaria en que se ha demostrado el papel de un microorganismo en su desencadenamiento. Esta enfermedad está producida por la reactividad cruzada entre los Ag presentes en la superficie del estreptococo del grupo A y Ag propios presentes en ciertos tejidos, como el corazón, las articulaciones, la piel, etc. La homología estructural entre estos Ag hace que los Ac producidos contra dicho microorganismo causen una reacción inflamatoria y daño de nuestros tejidos. En la psoriasis se ha descrito un aumento de linfocitos T específicos del estreptococo β-hemolítico y se ha propuesto que podría existir reactividad cruzada entre estos Ag y los presentes en la piel. También se ha propuesto que un traumatismo (fenómeno de Koebener), la administración de fármacos o la retirada rápida de tratamientos inmunosupresores pueden desencadenar la psoriasis6. En la artritis psoriásica se ha propuesto algún factor vírico7, aunque no se ha encontrado aún el factor ambiental causante del desencadenamiento de esta enfermedad.
Los factores ambientales desencadenarían la enfermedad en individuos genéticamente predispuestos. La susceptibilidad genética desempeña un papel muy importante en la patogenia de las enfermedades autoinmunitarias, y especialmente en la artritis psoriásica8. En consonancia, en la psoriasis y en la artritis psoriásica se observan una importante agregación familiar y una elevada concordancia de estas enfermedades entre gemelos monocigotos, que es del 65% para la psoriasis y del 30% para la artritis psoriásica9.
Aunque no existe duda sobre la importancia de la susceptibilidad genética, las enfermedades autoinmunitarias no se transmiten como enfermedades monogénicas clásicas, dominantes o recesivas, sino que son poligénicas, es decir, existen numerosos genes (en realidad, ciertos polimorfismos de estos genes) de susceptibilidad que actúan conjuntamente para producir una enfermedad. Estas variantes alélicas son normales en la población y por sí mismos no determinan si un individuo desarrollará o no la enfermedad (solo incrementan o disminuyen el riesgo de presentarla); solo cuando actúan conjuntamente con un factor ambiental se desencadenará la enfermedad. Como resultado, existe una considerable heterogeneidad genética y, por tanto, también clínica en estos pacientes, ya que no todos los pacientes van a compartir los mismos genes de susceptibilidad. Este es uno de los motivos por los que es difícil encontrar algún síntoma patognomónico en las enfermedades autoinmunitarias y se diagnostican frecuentemente utilizando criterios diagnósticos.
Implicaciones patogénicas de los estudios genéticosEn consonancia con un patrón de herencia poligénica se han descrito numerosos loci distribuidos a lo largo de todo el genoma humano asociados a su susceptibilidad. Los polimorfismos génicos asociados suelen estar implicados en el desarrollo de respuestas inmunitarias excesivas o inadecuadas. Por ello, la caracterización de los genes de susceptibilidad y de las respuestas implicadas tiene una gran importancia para descifrar la patogenia de esta enfermedad.
La principal asociación genética en la psoriasis y en la artritis psoriásica es con el alelo del gen de MHC de clase I, HLA-Cw*060210 (tabla 1). La función de estas moléculas es presentar péptidos derivados de las proteínas del citoplasma a los linfocitos T CD8. Cada individuo usualmente expresa diferentes alelos de las moléculas de MHC de clase I y cada uno de ellos presenta diferentes péptidos, lo que pretende evitar que algún patógeno pueda evadir el sistema inmunitario en todos los individuos de la especie. La susceptibilidad a las enfermedades autoinmunitarias está generalmente asociada a ciertos alelos de las moléculas de MHC, ya que solo algunos alelos van a ser capaces de presentar péptidos derivados de los auto-Ag responsables de la enfermedad. Esto sugiere que el papel de HLA-Cw*0602 en la patogenia de estas enfermedades sería probablemente la presentación de un auto-Ag citoplasmático presente en las células de la piel o la sinovia a los linfocitos T CD8 autorreactivos. En consonancia con un papel de los linfocitos T CD8 en la patogenia de la enfermedad, se ha descrito en la artritis psoriásica, pero no en la psoriasis, la asociación de genes, como MICA, que tiene un papel coestimulador en la activación de los linfocitos T citotóxicos11,12.
Genes/polimorfismos asociados a la susceptibilidad a la artritis psoriásica. En la tabla se muestran las asociaciones genéticas más relevantes descritas y su posible función en la patogenia de la enfermedad
| Gen | Cromosoma | Función |
| HLA-Cw*0602 | 6p21 | Presentar péptidos citoplasmáticos a linfocitos T CD8 |
| MICA | 6p21 | Molécula coestimuladora que tiene como ligando al receptor NKG2D expresado de linfocitos T CD8 y células NK |
| HLA clase II | 6p21 | Presentación de péptidos a los linfocitos T CD4 |
| IL12B | 5q33 | Subunidad de la citocina IL-23. La IL-23 está implicada en la generación y activación de los linfocitos Th17 |
| IL23R | 1p31 | Subunidad del receptor de la IL-23 |
| IL2/IL21 | 4q27 | La IL-21 es una citocina producida por los linfocitos Th17 |
| TNFA | 6p21 | Codifica la citocina inflamatoria TNF-α |
| TNFAIP3 | 6q23 | Codifica la enzima A20, implicada en la señalización de la respuesta al TNF-α |
| TNIP1 | 5q33 | Codifica para la proteína 1 de interacción con TNFAIP3. Ambas están implicadas en la señalización de la respuesta al TNF-α |
Diversos alelos de genes de moléculas MHC de clase II, como HLA–DRB1*04, HLA-DRB1*07, o más recientemente HLA-DR1713, también se han asociado a la susceptibilidad o las manifestaciones clínicas de la enfermedad. El hecho de que el papel de estas moléculas es la presentación de péptidos a los linfocitos T CD4 sugiere que estos linfocitos pueden también estar implicados en su patogenia.
La psoriasis también se asocia al alelo del gen TNFAIP3 y TNIP1, que codifican proteínas implicadas en la señalización del TNF-α6. TNFAIP3 está asociado a la artritis reumatoide y lupus eritematoso sistémico, aunque todavía no existen datos concluyentes sobre su papel en la artritis psoriásica. Sin embargo, en algunas poblaciones, la susceptibilidad a la artritis psoriásica se ha asociado con un polimorfismo del promotor del gen TNFA que favorece el incremento de la síntesis de TNF-α8.
Además, debemos destacar la asociación a esta enfermedad de diversos genes de citocinas implicadas en la respuesta Th176. La susceptibilidad a la psoriasis y a la artritis psoriásica se asocia tanto al gen que codifica para una de las subunidades de la IL-23 (IL12B), como al gen que codifica una de las subunidades de su receptor (IL23R). Esta citocina desempeña un papel clave en la generación de los linfocitos Th17. Una variante polimórfica cerca del gen IL23A (que codifica para la otra subunidad que forma la IL-23) también se asocia a la susceptibilidad a la psoriasis. Igualmente se ha asociado su susceptibilidad con una región génica localizada en el cromosoma 4q27, en la que se encuentra el gen de IL-21, citocina producida por las células Th17.
En conjunto, estos datos indican que genes de moléculas implicadas en la respuesta inmunitaria citotóxica, Th1 y Th17, están asociados a la susceptibilidad de la enfermedad, lo que sugiere que estas respuestas pueden tener un papel importante en su patogenia.
Características inmunopatológicas de la psoriasis y la artritis psoriásicaLa psoriasis y la artritis psoriásica comparten características histológicas e inmunológicas, lo que sugiere que comparten mecanismos patogénicos. Tanto en la piel como en las articulaciones se produce una reacción inflamatoria crónica caracterizada por un marcado incremento del número de vasos sanguíneos y de la infiltración de linfocitos T, macrófagos y neutrófilos activados.
En la psoriasis, la lesión de la piel se caracteriza por una hiperplasia epidérmica con queratinización excesiva, que da lugar a las características macroscópicas y microscópicas de la piel. Además, se observan una dilatación y un aumento del número de vasos sanguíneos, y una infiltración cutánea, sobre todo de linfocitos T, neutrófilos y macrófagos activados. Los linfocitos T CD8 infiltran mayoritariamente la epidermis y son clonales, lo que indica que pueden haber sido activados y expandidos en respuesta a un Ag. También hay un incremento de linfocitos CD4 policlonales en la dermis, que están polarizados mayoritariamente a Th1 y en menor medida a Th17. Los neutrófilos abundan en la dermis, donde pueden formar microabscesos, y parecen contribuir a la persistencia de la enfermedad.
La sinovitis en la artritis psoriásica muestra características similares a otras espondiloartropatías. Se caracteriza por una hiperplasia sinovial menos marcada que en la artritis reumatoide, una prominente vasculatura y un abundante infiltrado inflamatorio caracterizado por linfocitos T, neutrófilos y macrófagos activados. Hay un incremento selectivo de linfocitos T CD4 en la membrana sinovial y de CD8 en el líquido sinovial y hueso subcondral debajo de las entesis14–17. Igual que ocurre en la piel, los linfocitos T CD8 muestran una expansión clonal posiblemente en respuesta a un Ag. Los linfocitos T CD4 son policlonales y están polarizados fundamentalmente a Th1 y, en menor medida, a Th17. En consonancia con una respuesta Th1 y Th17, existe un incremento en la sinovia tanto de macrófagos activados que secretan TNF-α18 como de neutrófilos.
Con la evolución, la inflamación crónica puede causar la destrucción del cartílago y erosión ósea. Las erosiones son marginales, como en la artritis reumatoide, pero se vuelven irregulares con la progresión debido a la osteoclastogénesis y la neoformación ósea en la proximidad de las erosiones.
Mecanismos patogénicos implicados en el desarrollo de la artritis psoriásicaLa psoriasis y la artritis psoriásica son enfermedades muy complejas que comparten mecanismos patogénicos comunes. En estas enfermedades no se detecta la presencia de auto-Ac, descartando un papel destacado de la respuesta Th2 en su patogenia. Sin embargo, numerosas evidencias sugieren que tanto la respuesta inmunitaria citotóxica como la respuesta Th1 y Th17 pueden participar en el desencadenamiento y/o la perpetuación de la artritis psoriásica.
Existen diversas evidencias sobre el posible papel de los linfocitos T CD8 citotóxicos en la patogenia de la enfermedad. Primero, la asociación genética más importante en estas enfermedades es HLA-Cw*0602, cuya función es presentar péptidos citoplasmáticos a los linfocitos T CD8. Es importante destacar que HLA-Cw*0602 se asocia a un inicio precoz de la psoriasis y la artritis psoriásica19. Además, el fluido sinovial y las entesis de los enfermos están infiltrados por linfocitos T CD8 activados que parecen haberse expandido en respuesta a un Ag20. En la psoriasis, los linfocitos T CD8 predominan en la epidermis y son los primeros en infiltran la lesión psoriásica, lo que sugiere que pueden tener un papel inicial en el desencadenamiento de la enfermedad. Conjuntamente estos datos sugieren que la activación de linfocitos T CD8 autorreactivos puede desempeñar un papel clave en el desencadenamiento de la enfermedad, aunque esta afirmación debe ser tomada con cautela, ya que hasta la fecha no se conoce el auto-Ag responsable ni tampoco la célula que lo expresa.
También el TNF-α desempeña un papel importante en la patogenia de la enfermedad. Esta citocina es sintetizada mayoritariamente por los macrófagos y su función principal es inducir la inflamación. En las enfermedades autoinmunitarias inflamatorias, la activación crónica de los macrófagos por los linfocitos Th1 autorreactivos causa la secreción crónica del TNF-α y la perpetuación de la respuesta inflamatoria. Existen numerosas evidencias de que algo similar puede estar pasando en la artritis psoriásica. Como hemos descrito previamente, existe una significativa asociación del gen que codifica el TNF-α o de genes implicados en la respuesta a esta citocina con la susceptibilidad a la artritis psoriásica. Además, existe un aumento del número de linfocitos Th1, de macrófagos activados que sintetizan TNF-α y de diversas citocinas inflamatorias en la piel, sinovia y fluido sinovial18,21. Pero quizá la demostración más convincente es que el bloqueo de TNF-α con infliximab puede ser un tratamiento eficaz en esta enfermedad, que además puede disminuir algunos de los signos histopatológicos característicos de la artritis psoriásica, como la excesiva vascularización, la hiperplasia sinovial, la infiltración mononuclear y la activación de los osteoclastos, reduciendo la destrucción ósea22.
Por último, existen evidencias recientes que sugieren que los linfocitos Th17 y las citocinas producidas por estos linfocitos, como la IL-17, IL-21 e IL-22, también pueden causar inflamación y daño tisular en la artritis psoriásica y otras enfermedades autoinmunitarias23. La susceptibilidad genética a la psoriasis y a la artritis psoriásica se asocia con alelos de los genes que codifican para las cadenas de la IL-23 y su receptor6. Como hemos mencionado, esta citocina desempeña un papel clave en la generación y el mantenimiento de estas células. Además, las células Th17 están incrementadas en las articulaciones y en sangre, tanto en el inicio como en la progresión de la enfermedad24. Igualmente, hay un aumento de neutrófilos activados y de citocinas Th17, como la IL-17 y la IL-22, en la piel y la sinovia25–27. Por último, el tratamiento con un Ac monoclonal anti-IL-23 (ustekinumab) puede reducir los signos y síntomas de la enfermedad, lo que apoya un papel de IL-23 y de la respuesta Th17 en esta enfermedad28. A pesar de todas estas evidencias, todavía no se conocen cuáles son las bases moleculares y celulares que participan en esta respuesta. Además, se han descrito otros tipos celulares diferentes de los linfocitos Th17 capaces de producir IL-17 y que pueden participar en la patogenia de la enfermedad29. En conclusión, aunque parece que la esta respuesta puede desempeñar un papel destacado en la artritis psoriásica, sus bases moleculares y celulares todavía no han sido esclarecidas.
Integrando todos los mecanismos patogénicosTanto la respuesta citotóxica como Th1 y Th17 pueden intervenir en la patogenia de la artritis psoriásica, aunque desconocemos cómo se desencadena cada una de estas respuestas, cómo se desarrollan y cómo interactúan entre ellas (fig. 2). A partir de los datos anteriormente expuestos, podemos especular que los linfocitos T CD8 que se han expandido en respuesta a un Ag (propio o extraño) presentado en el contexto de las molécula HLA-Cw*0602 puede tener un papel en el desencadenamiento de la enfermedad. La respuesta mediada por linfocitos T CD8 autorreactivos produciría daño de las células sinoviales y la liberación de citocinas que puede favorecer el reclutamiento y la expansión de otras células inmunes como los linfocitos T CD4. El daño celular, la secreción de citocinas y el reclutamiento de células inmunitarias en la sinovia puede crear un caldo de cultivo adecuado para que en individuos genéticamente predispuestos se desarrollen otros mecanismos autoinmunitarios mediados por los linfocitos Th1 y/o Th17, que favorecerían el desarrollo de una respuesta inflamatoria crónica mediada por la activación de los macrófagos y los neutrófilos.
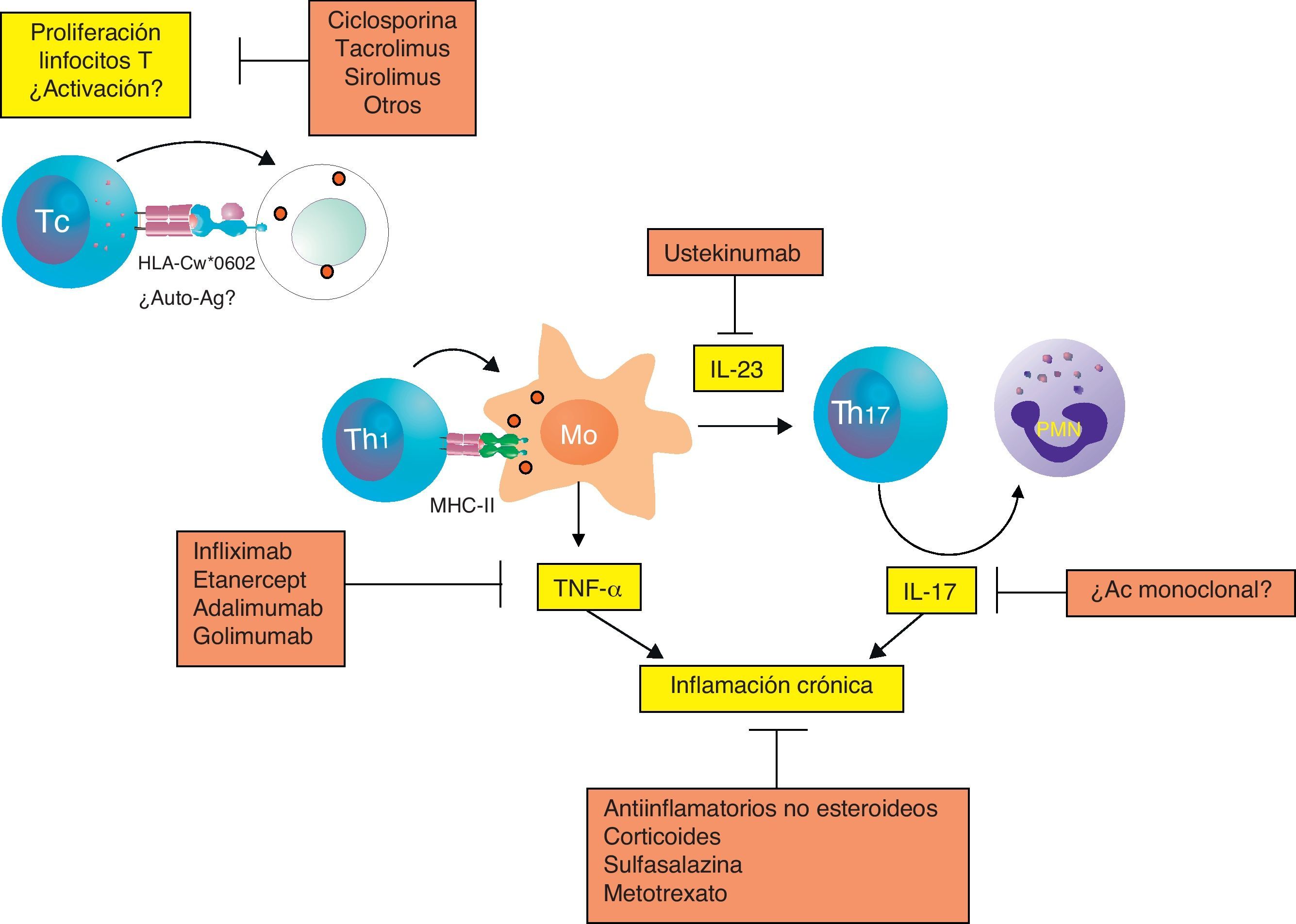
Modelo hipotético de los mecanismos implicados en la patogenia de la artritis psoriásica. Aunque no se ha desvelado completamente la patogenia de la enfermedad, las respuestas inmunitarias citotóxica, Th1 y Th17 parecen estar implicadas en el desencadenamiento y perpetuación de la artritis psoriásica. Los tratamientos actuales tratan de bloquear estos mecanismos inmunitarios que están desregulados en la enfermedad. MHC-II: MHC de clase II; Mo: macrófago; PMN: célula polimorfonuclear o neutrófilo; Tc: linfocito T citotóxico.
Lo que parece más obvio es que las citocinas que se producen en la respuesta Th1 y Th17 tienen una participación importante en el desencadenamiento de los síntomas y signos clínicos que observamos en los pacientes con artritis psoriásica. El TNF-α es uno de los principales responsables, ya que favorece la activación y el reclutamiento de células inmunitarias a la sinovia, favorece la hiperplasia sinovial, induce la secreción de metaloproteasas de matriz implicadas en la reabsorción osteoclástica del hueso y en la subsecuente degradación del cartílago, y junto con otros factores angiogénicos favorece la generación de nuevos vasos sanguíneos.
La liberación de citocinas generadas en la respuesta Th17 también participa en la patogenia de la artritis psoriásica. La IL-17 es una potente citocina inflamatoria destructora de las articulaciones. Es capaz de estimular la producción de IL-1 y TNF-α por los macrófagos, induce la secreción de IL-6 e IL-8 por los fibroblastos sinoviales y favorece el reclutamiento de neutrófilos y otras células inmunitarias a la sinovia. La IL-17 promueve la degradación del cartílago y también favorece la angiogénesis. Además, se ha sugerido que la IL-22 podría estar implicada en la hiperproliferación de los queratinocitos y de los sinoviocitos.
En la artritis psoriásica hay además osteodestrucción y neoformación ósea. El TNF-α, y posiblemente también la IL-1730, favorece la osteoclastogénesis de los monocitos y la subsecuente erosión ósea31. También existe neoformación ósea por mecanismos poco conocidos, aunque la utilización de inhibidores naturales sugiere que DKK-1 puede desempeñar un papel vital en la regulación de la remodelación ósea32.
De la patogenia al tratamientoLas enfermedades autoinmunitarias se pueden tratar inhibiendo la activación y la proliferación de linfocitos T autorreactivos. La ciclosporina, el tacrolimús (FK506) y el sirolimús (rapamicina) han sido ampliamente utilizados en el tratamiento de estas enfermedades porque inhiben específicamente la proliferación de los linfocitos T mediante el bloqueo de la síntesis o señalización de la citocina causante de su proliferación, la IL-2 (fig. 2). La ciclosporina ha sido utilizada más ampliamente y ha mostrado su capacidad de inducir una rápida mejoría de la psoriasis y una modesta mejoría de la artritis psoriásica. En los últimos años se ha avanzando mucho en el desarrollo de nuevos medicamentos que bloquean la activación de los linfocitos T y que han demostrado su potencial utilidad en algunas enfermedades autoinmunitarias. Desafortunadamente, todavía no han demostrado su eficacia en la artritis psoriásica.
Las enfermedades, como la artritis psoriásica, mediadas por linfocitos Th1 desencadenan reacciones inflamatorias crónicas muy intensas, que puedes ser tratadas con antiinflamatorios tanto esteroideos como no esteroideos, o con fármacos antirreumáticos como el metotrexato y la sulfasalazina. Estos tratamientos, aunque son útiles desde el punto de vista clínico, no atacan la causa de la enfermedad, y por tanto, tienen poca capacidad para afectar a la evolución o el daño de las articulaciones. Mucho más eficaz es bloquear directamente las moléculas responsables de esta utilizando antagonistas de TNF-α como infliximab, adalimumab, etanercept y golimumab.
Por último, la posible implicación de los linfocitos Th17 en la patogenia de la artritis psoriásica abre nuevas vías terapéuticas. Como hemos comentado anteriormente, el tratamiento con un Ac monoclonal anti-IL-23 (ustekinumab) puede reducir los signos y síntomas de la artritis psoriásica y apoya un papel de la IL-23 y de la respuesta Th17 en la artritis psoriásica. Existen otras citocinas que pueden participar en la diferenciación de las células Th17, como la IL-6. El bloqueo del receptor de la IL-6 con el Ac monoclonal tocilizumab podría tener una aplicación terapéutica que debe de ser evaluada. Igualmente, el bloqueo de IL-17 u otras citocinas implicadas en esta respuesta es otra potencial estrategia para el tratamiento de esta enfermedad que deberá ser evaluado en el futuro.
Es previsible que el completo esclarecimiento de la patogenia de esta enfermedad permita el desarrollo de nuevos tratamientos y una racionalización de los actuales, lo que previsiblemente redundará en un mejor pronóstico de los pacientes.
FinanciaciónEste trabajo ha sido financiado por Roche Pharma y por el proyecto del Fondo de Investigaciones Sanitarias (Instituto de Salud Carlos III) PS09/00420.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


