









This review emphasizes the significance of clinical findings, pathogenic mechanisms, diagnosis, management, and complications related to the ocular manifestations of rheumatoid arthritis, underscoring the need for interdisciplinary collaboration between rheumatologists and ophthalmologists.
MethodsA comprehensive literature search was conducted using PubMed, Embase, Web of Science, and Google Scholar for all English-language articles published from inception to July 2024. The following search string was utilized: (“rheumatoid arthritis”) AND (“dry eye disease” OR “dry eye” OR “episcleritis” OR “scleritis” OR “peripheral ulcerative keratitis” OR “central corneal ulcer*” OR “paracentral corneal ulcer*” OR “ocular manifestations” OR “ocular involvement”). Letters to the editor, editorials, short communications, case reports, case series, review articles, and original articles were reviewed, along with relevant references from those publications.
ResultsThe most common ocular manifestations in patients with rheumatoid arthritis include keratoconjunctivitis sicca (dry eye disease), episcleritis, scleritis, peripheral ulcerative keratitis, and central and paracentral corneal ulceration. Ocular involvement can be the initial presentation of the disease and may correlate with disease activity. If left untreated, ocular inflammation in rheumatoid arthritis can lead to permanent vision loss.
ConclusionsThe diverse ocular inflammatory manifestations in RA patients may indicate either the disease's initial presentation or the status of extra-articular activity. A multidisciplinary approach that includes rheumatologists and ophthalmologists is essential to prevent sight-threatening complications.
Esta revisión hace hincapié en la importancia de los hallazgos clínicos, los mecanismos patogénicos, el diagnóstico, el tratamiento y las complicaciones relacionadas con las manifestaciones oculares de la artritis reumatoide, subrayando la necesidad de una colaboración interdisciplinaria entre reumatólogos y oftalmólogos.
MétodosSe realizó una búsqueda bibliográfica exhaustiva utilizando PubMed, Embase, Web of Science y Google Scholar para todos los artículos en inglés publicados desde el inicio hasta julio de 2024. Se utilizó la siguiente cadena de búsqueda: («rheumatoid arthritis») AND («dry eye disease» OR «dry eye» OR «episcleritis» OR «scleritis» OR «peripheral ulcerative keratitis» OR «central corneal ulcer*» OR «paracentral corneal ulcer*» OR «ocular manifestations» OR «ocular involvement»). Se revisaron editoriales, comunicaciones breves, informes de casos, series de casos, artículos de revisión y artículos originales, junto con las referencias pertinentes de esas publicaciones.
ResultadosLas manifestaciones oculares más frecuentes en pacientes con artritis reumatoide son la queratoconjuntivitis seca, la epiescleritis, la escleritis, la queratitis ulcerosa periférica y la ulceración corneal central y paracentral. La afectación ocular puede ser la presentación inicial de la enfermedad y puede correlacionarse con la actividad de la enfermedad. Si no se trata, la inflamación ocular en la artritis reumatoide puede provocar una pérdida permanente de visión.
ConclusionesLas diversas manifestaciones inflamatorias oculares en pacientes con artritis reumatoide pueden indicar tanto la presentación inicial de la enfermedad como el estado de la actividad extraarticular. Un enfoque multidisciplinar que incluya a reumatólogos y oftalmólogos es esencial para prevenir complicaciones que pongan en peligro la visión.
Rheumatoid arthritis (RA) is a systemic autoimmune disease that affects the synovial membrane of joints.1 It predominantly occurs in female patients during the fifth and sixth decades of life. The results from the Global Burden of Disease Study 2019 estimated a global prevalence of 18.5 million cases.2 However, the prevalence of RA ranges from 0.31% to 0.9% in European countries,3 0.14% to 1% in Asia,4 0.6% to 0.9% in the United States and Canada, 2.7% among Australian indigenous populations,5 and 1.6% in Mexico.6
Extra-articular manifestations in RA, observed in 17.8–40.9% of cases, may affect the heart, central and peripheral nervous system, lungs, kidneys, gastrointestinal tract, and eyes, among others. These manifestations are often associated with a more severe course, higher mortality and morbidity, and positive titers of autoantibodies such as rheumatoid factor (RF) or anti-citrullinated protein antibody (ACPA).7
RA is the most common rheumatic disease associated with ocular involvement, presenting in 25–39% of patients. The duration of RA increases the risk of ocular involvement.8,9 Keratoconjunctivitis sicca (KCS) is the most frequent ocular manifestation, reported in 15–28% of patients. KCS is usually mild, and its course is independent of RA disease activity.10,11 On the other hand, secondary Sjogren syndrome (sSS), a condition that develops in patients with autoimmune diseases, including RA, represents a severe form of KCS that may be sight-threatening and thus requires aggressive management. Other potentially sight-threatening extra-articular manifestations include diffuse anterior and necrotizing scleritis (NS), peripheral ulcerative keratitis (PUK), and central and paracentral corneal ulceration (CPCU).12–14
Prompt identification of ocular manifestations in RA is necessary for preserving vision and quality of life. This comprehensive review provides an in-depth analysis of the epidemiology, pathogenic mechanisms, clinical presentation, diagnosis, and treatment options for ocular manifestations associated with RA.
Extra-articular manifestations of rheumatoid arthritisCardiovascular disease (CVD) remains the leading cause of death among patients with RA, presenting a two-fold risk of myocardial infarction and a 50% higher mortality risk from CVD compared to the general population.15–17 Pulmonary involvement accounts for the second most common (30%–40%) cause of death.18 Clinically significant interstitial lung disease develops in 8% to 15% of RA patients, accounting for 20% of all-cause mortality associated with RA.19 Atlantoaxial involvement is the most frequent (43–86%) neurologic manifestation of RA, occurring in 43–86% of patients, while rheumatoid nodules are the most common skin finding, affecting 30–40% of those with RA.20 Psychiatric comorbidities, such as depression (20%) and anxiety (70%), are also prevalent among RA patients.8,21
Hepatic disease, which manifests as drug hepatotoxicity, viral hepatitis, and fatty and autoimmune liver disease, occurs in 44% of RA patients.22 Dysphagia can result from xerostomia-related oropharyngeal dysmotility in patients with sSS, amyloidosis, or nerve compression from atlantoaxial subluxation.8,23 Notably, inflammatory eye manifestations occur in approximately 20% of RA patients.10 Dammacco et al. reported a 29% prevalence of KCS, 10% of episcleritis, 6% of anterior uveitis, 5% of scleritis, and 2% of PUK.24 The relevance of ocular manifestations is significant as they may be the initial presentation of the disease in up to one-third of the cases and may reflect an occult systemic disease activity. Furthermore, a severe dry eye disease (DED) associated with SS, NS, and destructive ulcerative keratitis leads to considerable visual morbidity, resulting in a detrimental quality of life for the patients.
Ocular manifestations and pathogenic mechanisms of rheumatoid arthritis (Fig. 1)The tear film and the ocular surfaceThe tear film is a complex dynamic mucin-aqueous gradient with an apical bipolar lipid layer multifunctional fluid that forms the interface between the environment and the ocular surface epithelia. The conjunctival epithelial cells produce transmembrane mucins, while the goblet cells produce water-soluble gel-forming mucins. Transmembrane mucins form the innermost layer of the tear film, facilitating the anchorage of the aqueous phase to reduce the tear film's superficial tension while the aqueous soluble mucins spread through the aqueous phase.25 The accessory and main lacrimal glands produce the tear film's middle (aqueous) layer. It comprises electrolytes, secretory IgA, lactoferrin, lysozymes, and trophic and wound healing factors.26 The meibomian glands in the eyelids secrete the oily phase or meibum, which forms the lipid outer layer of the tear film.27 It comprises an inner polar layer interacting with the aqueous layer and a thicker nonpolar layer facing the air. The amphiphilic properties of the polar layer create an interphase between the nonpolar layer and the mucin/aqueous phase, helping to stabilize the tear film.

On the other hand, the nonpolar layer prevents tear evaporation, creates a physical barrier that protects the ocular surface from the external environment, and reduces the friction forces created by constant blinking.28 Meibomian gland dysfunction (MGD) typically results from inflammatory blockage of the terminal glandular excretory ducts, causing tear film instability and evaporative DED. MGD is closely linked to increased RA disease activity scores.29 This association between MGD and RA is supported by studies indicating that women with RA frequently exhibit androgen deficiency, a condition that significantly elevates the risk of MGD. Additionally, the theoretical release of pro-inflammatory cytokines in the tear film may impact the terminal ducts of the meibomian glands, contributing to MGD in the context of heightened inflammation in RA patients. However, the underlying physiology of this phenomenon remains to be fully understood.30
Adequate tear production and blinking reflexes, regulated by the afferent sensory innervation supplied by the ophthalmic branch of the trigeminal nerve, are essential for maintaining the integrity of the ocular surface and tear clearance.26 Bitirgen et al. found that increased disease activity in patients with RA was significantly associated with corneal nerve fiber loss and reduced corneal sensitivity.31 Thus, inadequate inflammatory control leading to reduced corneal sensitivity in RA patients may result in incomplete blinking, MGD, and evaporative DED. Furthermore, the loss of nerve fibers could also be secondary to a vasculitic process that affects the vasa nervorum, as seen in RA-associated neuropathy, resulting in the infarction of individual peripheral nerves.32Table 1 illustrates the epidemiologic characteristics of significant clinical studies reporting on RA-associated ocular complications.33–64
Epidemiologic characteristics of the most relevant clinical studies including patients who developed RA-associated ocular complications.
| Author (year) | Country | Type of study | Total no. patients | Sex (F/M) | Mean age (years)a | Ocular complications (%) |
|---|---|---|---|---|---|---|
| Reddy et al. (1977)33 | India | Observational | 100 | 64/36 | N/A | KCS (22), cataract (9), anterior uveitis (4), scleritis (1), disk edema (1), episcleritis (1) |
| Bhadoria et al. (1989)34 | India | Observational | 107 | N/A | N/A | KCS (17.7), episcleritis (0.9) |
| Andonopoulos et al. (1989)35 | Greece | Observational | 111 | N/A | N/A | sSS (30.6) |
| Reddy et al. (1996)36 | India | Observational | 325 | 248/77 | 34.7 | KCS (16.6), episcleritis (3.7), scleritis (0.9), PUK (0.9), anterior uveitis (0.3) |
| Averns et al. (1999)37 | UK | Cross-sectional | 340 | N/A | N/A | KCS (37.4) |
| Uhlig et al. (1999)38 | Norway | Cross-sectional | 636 | 510/126 | 53.6 | KCS (37.6) |
| Cimmino (2000)39 | Italy | Observational | 587 | 451/136 | 58.9 | KCS (17.5) |
| Gilboe et al. (2001)40 | Norway | Case control | 81 | 72/9 | 44 | KCS (33.3) |
| Shaw et al. (2003)41 | India | Observational | 54 | N/A | N/A | DED (5.6), episcleritis (3.7) |
| Brun et al. (2003)42 | Norway | Cohort | 56 | 38/18 | 63a | KCS (51.8) |
| Carmona et al. (2003)43 | Spain | Observational | 788 | 562/226 | 48 | sSS (17), episcleritis and scleritis (2.5) |
| Turesson et al. (2003)44 | USA | Observational | 609 | 445/164 | 58.1a | KCS (10), sSS (9.5), episcleritis (0.8), scleritis (0.7) |
| Fujita et al. (2005)45 | Japan | Case control | 72 | 67/5 | 64 | sSS (9.7), KCS (91.7) |
| Calguneri et al. (2006)46 | Turkey | Observational | 526 | 453/73 | 48 | KCS (11.4) |
| Punjabi et al. (2006)47 | India | Case Control | 84 | 54/30 | N/A | DED (27.3) |
| Bettero et al. (2008)48 | Brazil | Observational | 198 | 169/29 | 51.6 | KCS (16.2), sSS (11.1), scleritis (2), PUK (2) |
| Hochberg et al. (2008)49 | USA | Observational | 16,752 | 12,061/4691 | 59.8 | KCS (4), anterior uveitis (0.9), episcleritis/scleritis (0.5) |
| Zlatanovic et al. (2010)50 | Serbia | Observational | 691 | 601/90 | N/A | KCS (17.7), episcleritis (5.1), scleritis (2), keratitis (1.6) |
| Kobak (2011)51 | Turkey | Observational | 165 | 125/40 | 52.5 | KCS (40.6) |
| Markovitz et al. (2011)52 | Israel | Observational | 61 | 55/6 | 51.9 | DED (85) |
| Eser et al. (2012)53 | Turkey | Observational | 150 | 126/24 | 53.2 | KCS (12) |
| Kouakou et al. (2014)54 | Ivory Coast | Observational | 24 | N/A | N/A | Cataract (12.5), keratitis (8.2), anterior uveitis (4.2) |
| Vignesh & Srinivasan (2015)55 | India | Observational | 196 | 150/46 | 43.9 | DED (28), filamentary keratitis (3), episcleritis (3), scleritis (2), cataract (1), PUK (1) |
| Domngang Noche (2016)56 | Cameroon | Observational | 19 | 16/3 | 60 | KCS (25), cataract (25), glaucoma (25) |
| Aboud et al. (2017)57 | Egypt | Observational | 180 | 150/30 | 45 | KCS (28.9), scleritis (1.7), episcleritis (1.7), keratitis (1.7) |
| Caimmi et al. (2018)58 | USA | Cohort | 92 | 63/29 | 62 | Scleritis (35.9), episcleritis (25), PUK (22.8), anterior uveitis (15.2) |
| Akintayo et al. (2019)59 | Nigeria | Observational | 50 | 42/8 | 47.2 | KCS (30), cataract (26), scleritis/episcleritis (8), disk edema (8), glaucoma (6), PUK (2) |
| Guellec et al. (2020)60 | France | Cohort | 811 | 622/189 | 47.7 | KCS (28.4) |
| Uribe-Reina et al. (2021)61 | Colombia | Observational | 266 | N/A | N/A | KCS (12.8), scleritis (2.6), uveitis (1.1), keratitis (0.8), PUK (0.4), episcleritis (0.4) |
| Cetinkaya (2022)62 | Turkey | Observational | 2691 | 2067/624 | NR | DED (26), episcleritis (4.6), scleritis (1.4) |
| Lai et al. (2023)63 | Taiwan | Case control | 33,398 | 22,092/11,306 | 49.6 | DED (16.7), sSS (6.2), corneal ulcer (1.2) and opacity (0.7) |
| Baquero-Ospina et al. (2023)64 | Mexico | Observational | 79 | 73/6 | 54.9 | KCS (62), scleritis (22.8), PUK (12.7), retinal vasculitis (1.3), anterior uveitis (1.3) |
KCS, or DED, is characterized by inadequate ocular surface lubrication due to decreased tear production, rapid evaporation, or both.65 KCS is diagnosed based on symptoms of dry eye and clinical signs. In contrast, the diagnosis of Sjögren's syndrome relies on characteristic clinical signs and symptoms, supported by specific tests such as salivary gland histopathology and the presence of serum autoantibodies. Additionally, the American European Consensus Group (AECG), composed of the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR), classification criteria serve as valuable tools that aid clinicians in making a diagnosis.66 SS can be classified as either “primary”, when it occurs as an isolated condition, or “secondary”, when it is associated with a systemic inflammatory disease such as RA, systemic lupus erythematosus (SLE), or systemic sclerosis.67 More recent publications suggest using the term “associated” rather than “secondary” to reflect the reciprocal influence of SS and the underlying systemic condition on each other's clinical manifestations, laboratory findings, and outcomes.68 In both clinical scenarios, lymphocytic infiltration of the productive acini of the lacrimal and salivary glands results in KCS and xerostomia, respectively.69 Interestingly, certain clinical features—including parotid gland enlargement, lymphadenopathy, purpura, Raynaud's phenomenon, renal involvement, and myositis—have been reported more frequently.70
Epidemiologic featuresDED is the most common ocular manifestation of RA, while RA is among the most prevalent systemic autoimmune diseases linked to DED.65 The incidence of DED in the general population ranges from 5% to 17%, while in RA, it is reported to be between 19% and 31%.71 Among patients with RA and sicca signs and symptoms, up to 10% fulfill the sSS diagnostic criteria.72 A study reported sSS developing in 36% of the eyes five years after RA was diagnosed, 23% between 5 and 10 years, and 41% after ten years.73
PathogenesisThe pathogenesis of sSS related to RA remains poorly understood. The histopathology of lacrimal glands is characterized by persistent lymphocytic infiltration and eventual acini destruction, indicating a potential common pathway in the pathogenesis of DED in RA and other autoimmune diseases.72 There is T-cell-mediated activation of inflammatory mediators (interleukin [IL]-1, IL-17, tumor necrosis factor [TNF]-α) in RA patients’ joints and the ocular surface. These cytokines create an inflammatory environment that promotes oxidative stress, disrupting the immunological tolerance of the lacrimal gland and ocular surface epithelia.72 In patients with RA-associated sSS, Villani et al. reported that tear fluid concentrations of IL-1α and IL-6 significantly decreased after treatment.74 In RA patients without sSS, there were no changes in the pro-inflammatory cytokine milieu after treatment, suggesting a dissimilar ocular surface pathology between RA patients with and without sSS.74 Elevated levels of IL-17 have also been reported in the tears of RA patients with KCS compared to healthy controls.75
In patients with SS, including those associated with RA, inflammation of the lacrimal gland results in reduced tear production and DED.76 Additionally, there is a statistically significant association (p=0.016) between elevated tear osmolarity (>316mOsm/L), a surrogate marker for ocular inflammation and DED, and increased disease activity score 28-erythrocyte sedimentation rate (DAS28-ESR) values.77
Marsovsky et al. found an increased density and activation of Langerhans cells (LCs) in the cornea of RA patients without evident ocular surface inflammation; however, this was associated with hypo-lacrimation severity. LCs, situated in the peripheral superficial cornea, are known to regulate the early adaptive and innate immune responses on the ocular surface.78 IL-1 inhibitors, anti-TNF-α biologic agents, and systemic corticosteroids inhibit the in vitro migration of LCs, thereby maintaining ocular immune privilege and promoting anterior chamber-associated immune deviation.79 Usuba et al. observed improvements in tear production and the recovery of conjunctival goblet cells in RA patients treated with anti-TNF-α therapy.29
Clinical manifestationsSymptoms include ocular discomfort, dryness, redness, burning and foreign body sensation, fluctuating and blurry vision, and pain.80 Conversely, signs can consist of varying degrees of conjunctival hyperemia, decreased tear production, increased evaporation rate, and staining of the conjunctiva and cornea.71,81 Depending on the severity of dryness, the cornea may range from mild zonal superficial punctate keratitis to more severe conditions like diffuse superficial punctate keratitis, patchy epitheliopathy, filamentary keratitis, and epithelial defects.82
DiagnosisWhile minor changes in the ocular surface require a slit-lamp examination by an eye care specialist, an initial dry eye assessment can be conducted by rheumatologists. The Ocular Surface Disease Index (OSDI) questionnaire assesses dry eye symptoms, environmental triggers, and changes in visual function. It has been validated in multiple languages and can be utilized by non-ophthalmologists.83 The Schirmer-I test measures basal and reflex tear secretion.13 This test involves placing a 35mm long filter paper strip in the outer commissure of both lower eyelids and waiting five minutes to assess the wet portion. To minimize excessive reflex stimuli, conduct the test in a dimly lit room and instruct the patient to remain silent with their eyes closed. A wetting measurement greater than 10mm in 5min indicates a normal result, while measurements below 5mm and between 5mm and 10mm suggest severe and moderate dry eye, respectively.84
Ng et al. reported a significantly lower Schirmer score (4.5±2.3mm) in RA patients with associated SS compared to those without it (16.4±10.0mm).85 Moreover, patients with SS secondary to RA have been reported to exhibit more severe superficial punctate keratitis, higher ocular surface staining (OSS) scores, lower fluorescein tear-film breakup time (FTBUT), and lower Schirmer scores compared to those with primary SS (pSS) and SS secondary to SLE.13 The sensitivity and specificity of the Schirmer I test in SS are fairly high. According to the European Community Study Group on Diagnostic Criteria for Sjögren's Syndrome, the sensitivity and specificity of the Schirmer test, among patients with pSS and sSS, was 77% and 72%, respectively.86 While the Schirmer test is simple and easily accessible for non-ophthalmologists, its results must be interpreted cautiously due to their low reproducibility.87
The OSS, also known as the SICCA score, is a quantitative measurement of DED-associated ocular surface damage. It uses a combination of vital dyes to stain the damaged corneal (fluorescein 0.5%) and conjunctival (lissamine green 1%) epithelia, with an eye care specialist observing the staining patterns at the slit lamp (Fig. 2).81 Receiver operating characteristic curves have shown optimal cutoff values of five points for the OSS, with a sensitivity of 56% and a specificity of 75% (Youden's J=0.31). However, although OSS staining scores are helpful for SS research classification, they are subject to significant interrater and intrasubject variability.88 In a cohort of 38 RA patients with moderate to severe DED, but without sSS, Yu et al. reported significantly higher corneal fluorescein staining values (4.2±3.4 vs. 3.2±2.7, p=0.02) and higher composite dry eye severity score based on signs (0.47±0.14 vs. 0.42±0.12, p=0.002) compared to non-RA patients with moderate to severe DED.89 Lissamine green staining scores were also high, but without reaching statistical significance (3.0±1.8 vs. 2.6±1.5, p=0.06).89

Ocular surface staining score (OSS) from the Sjögren's International Collaborative Clinical Alliance (SICCA) in an RA patient with severe dry eye disease due to secondary Sjögren syndrome. (A) Nasal conjunctival lissamine green staining (3 pts); (B) Corneal fluorescein staining (3 pts)+1 pt for coalescence+1 pt for pupillary affection (total=5 pts); (C) Temporal conjunctival lissamine staining (2 pts). OSS total score=10pts.
FTBUT, which may be evaluated with fluorescein staining or non-invasive methods, was significantly lower in RA patients than in controls. In contrast, the Meibomian gland function, corneal fluorescein staining, and eyelid margin were also worse in such patients.71 Studies have reported that RA patients with sSS had significantly shorter FTBUT and higher corneal staining scores and even increased CRP and ESR rates than non-sSS patients.85
TreatmentFrequent instillation of preservative-free, hypotonic, and high colloidal osmolality lubricant eyedrops containing hyaluronic acid, glucans, or methylcellulose are ideal for the treatment of sSS-DED associated with RA.90,91 Higher viscosity and mucoadhesive formulations have a longer retention time, generating more relief but may cause temporary blurred vision. Carboxymethyl cellulose (0.25–1%) and sodium hyaluronate (0.1–0.4%) may be prescribed 4 times daily for a few months in mild DED.92,93 In severe cases, high-viscosity gels such as carbomer 0.4% may be prescribed four times daily for several months.92 Topical immunomodulatory drugs (cyclosporine 0.05%, tacrolimus 0.03%, lifitegrast 5%) are now considered first-line therapy for autoimmune inflammatory-related DED. They may be accompanied by short courses of surface corticosteroids, including hydrocortisone 0.33% or secretagogues (diquafosol tetrasodium 3%).94–98 For more severe cases, interventional therapies, including bandage and scleral contact lenses (prosthetic replacement of the ocular surface ecosystem [PROSE]), punctal occlusion, tarsorrhaphy (partial or complete surgical closure of the upper and lower eyelid), and salivary gland transplant are necessary to improve the condition.83,99
ComplicationsImpaired ocular surface lubrication significantly increases the risk of corneal and conjunctival infections in patients with SS. The latter occurs due to the absence of lacrimal antimicrobial proteins from the innate immune system, including secretory IgA and lysozyme lactoferrin. In a study of 67 women with DED, including 29 with SS, Hori et al. reported a significantly higher rate of fluoroquinolone-resistant methicillin-sensitive and methicillin-resistant coagulase-negative Staphylococcus compared to controls.100 In another study of 66 patients with microbial keratitis, Jhanji et al. found that the prevalence of RA and DED was 81% and 26%, respectively. The administration of corticosteroids for dryness in RA patients may contribute to the increased risk of infection.101 Antimicrobial peptides, such as cathelicidin LL-37 and human β-defensins, also provide innate protection to the ocular surface and can be upregulated in patients with DED.102 In fact, DED-associated disruption of the corneal barrier function and homeostasis may result in corneal epithelial erosion, ulceration, stromal melting, and perforation.63 A large nationwide matched cohort study of 33,398 patients with RA and 33,398 non-RA patients reported a statistically significant incidence per 1000 person-years of corneal ulcers in the former group (1.51 vs. 1.14, p=0.0009).63 In Sections “Peripheral ulcerative keratitis (PUK) and Central and paracentral corneal ulceration (CPCU)”, we will discuss in detail the association between corneal ulceration with PUK and CPCU.
EpiscleritisDefinition and backgroundEpiscleritis is typically an idiopathic, benign, self-limiting condition resulting from inflammation of the episclera. It causes localized segmental redness, photophobia, discomfort, hypersensitivity to touch, and mild pain.55,103
Epidemiologic featuresThe prevalence of RA-associated episcleritis is 1–5%.55 Following KCS, it is the most common ocular manifestation associated with RA.9 According to Watson, episcleritis is classified into simple and nodular forms.103 The former type is the most common (up to 78% of cases), involving a larger area of the episcleral surface. In contrast, the latter is characterized by an elevated, localized nodular formation on the episclera.104 Sainz de la Maza et al. reported that episcleritis was primarily unilateral (60%) and more common in women (64%). Furthermore, it occurred at a younger age (48 vs. 54 years) and was less frequently associated with ocular complications than scleritis.104
PathogenesisEpiscleritis is a non-granulomatous inflammation affecting the episclera's superficial vessels and vascular plexus. This condition results in vasodilation, increased permeability, and perivascular infiltration of macrophages and white blood cells.105 The pathogenic mechanisms underlying episcleritis remain unknown. However, Bhamra et al. hypothesized that it may result from systemic inflammatory signaling leading to dysregulation of the tear cytokine milieu (i.e., transforming growth factor [TGF]-β and IL-1α) with subsequent loss of tear integrity.9 The latter, in turn, resulting in episcleritis. The inflammatory process is acute, self-limited, and may last up to three weeks.106 Nodular episcleritis may have a more recurrent presentation pattern and is more frequently associated with a systemic disease, including RA.103
Clinical manifestationsDuring episodes of inflammation, the superficial vessels of the episcleral plexus become congested and are easily seen as a flat sectoral redness under the slit lamp.9 The nodular variant is an elevated nodule featuring movable edema and infiltration (Fig. 3).107

Episcleritis in patients with early diagnosis of Rheumatoid Arthritis. (A) Acute simple episcleritis on the temporal quadrant of the left eye showing superficial ingurgitated vessels of the episcleral plexus. (B) Same eye after 3min of 5% phenylephrine eye drop instillation, showing significant vessel blanching. (C) Active recurrent nodular episcleritis in a young patient with unknown RA.
Since tortuous superficial vascular networks characterize episcleritis, vessels typically blanch after the application of phenylephrine hydrochloride 2.5% (Fig. 3). The hindmost is not present in scleritis (positive phenylephrine test).108 Watson and Hayreh reported that patients with simple episcleritis exhibited localized and diffuse redness with or without grayish deposits in 69% and 31% of cases, respectively. Eye tenderness was observed in 33% of simple episcleritis cases.103 In contrast, nodular episcleritis was marked by localized redness in 85% of cases and widespread redness in 15%. The nodule in these patients was mobile, without scleral involvement, and surrounded by congestion.103
TreatmentA study reported that in 17% of episcleritis patients, the inflammation subsided without treatment, while 50% required topical corticosteroids (fluorometholone QID), and 17% oral non-steroidal anti-inflammatory drugs (NSAIDs) (25mg indomethacin QID) to control the inflammation. Patients unresponsive to fluorometholone were switched to prednisolone acetate, a higher-potency topical corticosteroid.109
ComplicationsEpiscleritis complications are rare, with most cases resolving within two to three weeks without treatment or lasting effects. Sainz de la Maza et al. reported an 11% prevalence of anterior uveitis, 4% glaucoma, and 2% cataracts among 94 patients with episcleritis of any cause.110
ScleritisDefinition, background, and classificationScleritis is a severe and painful scleral stroma inflammation that can lead to irreversible vision loss.104 It is anatomically divided into anterior scleritis, which affects the area anterior to the extraocular muscles, and posterior scleritis, situated posterior to the extraocular muscle insertion.103 While anterior scleritis appears as an injected sclera, with or without corneal inflammation and anterior chamber cells, posterior scleritis is characterized by choroidal folds, optic disk swelling, serous retinal detachment, and a T-sign on B-mode ultrasound.9 Anterior scleritis is subdivided into diffuse, nodular, and necrotizing, with or without inflammation.111 Because each scleritis subtype has different prognostic and treatment implications, identifying them is crucial for the outcome.
Histologic studies performed on severe scleritis specimens found a vasculitic response characterized by vessel thrombosis, neutrophil infiltration, immune-complex deposition, and fibrinoid necrosis of the vessel walls.108,112 The conjunctival epithelium exhibited diffuse staining for human leukocyte antigen (HLA)-DR glycoproteins, whereas normal epithelium does not. Also, the substantia propria and sclera show significantly higher numbers of macrophages, B-cells, HLA-DR glycoproteins, T cytotoxic/suppressors, and T-helper cells than healthy controls.112 Moreover, according to the criteria developed by Scott and Bacon, scleritis associated with atypical digital infarcts or histopathologic evidence of vasculitis makes a diagnosis of rheumatoid vasculitis most probable, indicating a potentially deadly condition.113
Epidemiologic featuresRA is the most common systemic cause of scleritis.114 In a large cohort of 500 scleritis cases, the mean age at presentation was 53.7 years (ranging from 12 to 96 years), with a female preponderance of 71% and unilateral disease in 58% of cases. RA was the most frequently associated systemic disease (6.4%).104 The prevalence of diffuse, nodular, necrotizing, and scleromalacia perforans was 75%, 14%, 4%, and 1%, respectively. Posterior scleritis accounted for only 6% of cases.104 However, when comparing the clinical characteristics of idiopathic and RA-associated scleritis, the latter occurred in older patients (61 years), was more frequently necrotizing (34% vs. 3%), more often bilateral (53% vs. 34%), resulted in vision loss (59% vs. 20%), and led to PUK development (31% vs. 0%).115 Another study reported that scleritis onset was delayed, occurring 15 years after the diagnosis of RA was established,58 contrasting with granulomatosis with polyangiitis (GPA), where scleritis tends to develop earlier or as the first manifestation of the disease.116 This finding was also noted by Akpek et al. who reported that scleritis occurred after an established diagnosis of RA in 33/37 (89%) of cases, and it occurred in only 13/22 (59%) of cases that were ultimately diagnosed with a systemic vasculitis such as GPA, polyarteritis nodosa (PAN), Takayasu arteritis, among others.117 Sainz de la Maza et al. reported that patients with scleritis associated with GPA had significantly more NS, decrease in vision, and PUK compared with patients with RA-associated scleritis.118 However, several features of systemic vasculitis have been found in tissues of patients with RA-associated NS (see Section “Pathogenesis”). In fact, rheumatoid vasculitis, which may present decades after inactive RA, is part of the 2012 Chapel Hill Consensus Conference nomenclature.119,120
PathogenesisNo other ocular manifestation related to RA shares as much pathogenic similarity with its primary manifestation as scleritis. The unique histologic structure of the sclera, characterized by its collagen content, likely plays a crucial role in its pathogenic origin. Like RA arthritis, scleritis may be caused by the deposition of immune complexes, leading to microvasculitis of capillaries and arterioles. Ultimately, the phagocytosis of immune complexes and the release of inflammatory cytokines contribute to scleral damage.14,112,115 Enucleated globes from RA necrotizing scleritis exhibit granulomatous inflammation with central necrosis, surrounded by an external infiltration of plasma cells and lymphocytes and an internal infiltration of neutrophils and histiocytes.121 Biopsies of the sclera and conjunctiva from RA patients demonstrated C1q immune complex deposition in 40% of cases. Immunofluorescence (IF) analysis revealed C3 with IgA, C4 with IgA, and IgG, each present in one case within the scleral vasculature.112 Furthermore, treatments targeting macrophages, IL-4, TNF-α inhibition, and B-lymphocytes have demonstrated greater efficacy in murine models and case reports of RA patients with scleritis.122 TNF-α and IL-1 are well-identified cytokines in the pathogenesis of RA, necrotizing scleritis, and PUK (see Section “Peripheral ulcerative keratitis (PUK)”), likely associated with the increased production of MMPs in these three conditions.9
Clinical manifestationsPatients may experience notable redness and intense pain, often described as sharp, stabbing, or a sensation of fullness that worsens at night.9 During the slit-lamp examination, scleral edema is observed by the bowing of the posterior edge of the slit light beam and engorgement of deep scleral plexus vessels in a crisscross pattern, resulting in a salmon-red appearance of the eye. The phenylephrine test will not induce blanching of the deeply congested vessels in the eyes affected by scleritis.108 NS is characterized by thinning and a “porcelain white” ischemic appearance of the necrotic sclera. The uveal show displays a violaceous or black hue through the translucency of the sclera, indicating severe scleral thinning (Fig. 4).111
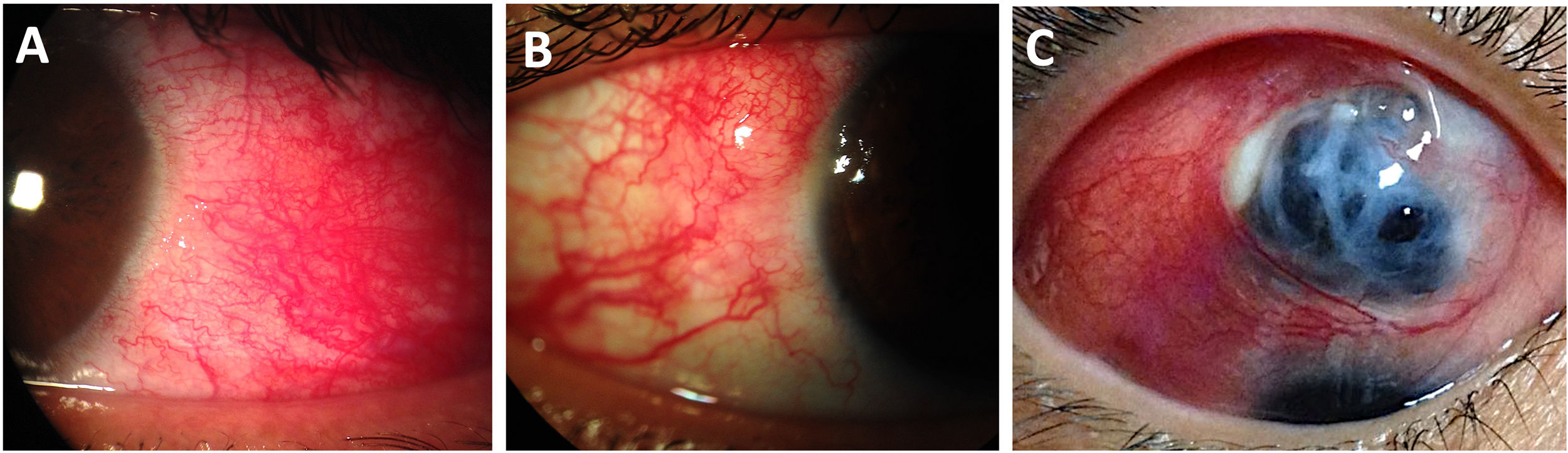
Scleritis in patients with longstanding Rheumatoid Arthritis. (A) Anterior diffuse scleritis on the temporal quadrant of the left eye showing deep dilated vessels with scleral edema. (B) Active nodular scleritis in the superior temporal area of the right eye. (C) Extensive necrotizing scleritis with significant uveal bulge surrounded by ischemic sclera and deep scleral vasculature ingurgitation.
As stated before, RA is the most common systemic disease associated with scleritis.123 The diagnosis of anterior scleritis is determined through direct visualization, while the posterior counterpart is diagnosed by combining B-mode ultrasonography with clinical findings. These findings include visual distortion linked to choroidal folds, retinal striae, serous retinal detachment, associated anterior scleritis, and optic disk swelling.124 The T-sign is the most frequent B-mode ultrasound finding in posterior scleritis, resulting from fluid accumulation behind Tenon's capsule.124 In cases of diagnostic uncertainty, a CT scan or MRI may reveal eccentric thickening and enhancement along with focal peri-scleral cellulitis.125 Infectious scleritis characterized by marked lid swelling and mucopurulent discharge must always be ruled out.111 Serological testing for tuberculosis and syphilis must also be obtained. The sclera and cornea, if involved, can be scraped for smear and cultures in eyes with signs of infection.111
TreatmentRA-associated scleritis requires a combined treatment approach that includes systemic immunosuppression with corticosteroids, steroid-sparing conventional immunosuppressive drugs like methotrexate, azathioprine, mycophenolate mofetil, and leflunomide. In severe cases, the regimen may add biologic agents, such as rituximab, anti-TNFα agents, and cyclosporine A or cyclophosphamide.109,111,126–128 Patients with scleritis who have an associated systemic disease, such as RA, are less likely to respond to systemic NSAIDs.129 On the other hand, patients with nodular or diffuse scleritis with an associated systemic disease showed a significant response to immunosuppressive therapy (IMT).129 Methotrexate (MTX) is the first-line agent for managing RA and can, therefore, be used in cases of associated scleritis. However, alkylating agents are the most effective immunosuppressive drugs for diffuse or nodular scleritis, especially in cases involving potentially lethal associated systemic disease and necrotizing scleritis, particularly those linked to small-vessel granulomatous vasculitis (i.e., GPA, PAN, and micro-PAN).129
ComplicationsA study reported a 37% prevalence of visual loss, defined as a decrease in vision of ≥2 Snellen lines in patients with scleritis compared to 2% in episcleritis.110 Comparing the prevalence of ocular complications between RA-associated scleritis and idiopathic scleritis, or scleritis associated with other systemic autoimmune diseases, necrotizing scleritis (34% vs. 3%), vision loss (59% vs. 20%), and PUK (31% vs. 0%) were significantly more common in RA-scleritis cases compared to idiopathic ones. Conversely, no significant difference was observed when considering other systemic autoimmune diseases.130
Peripheral ulcerative keratitis (PUK)Definition and backgroundPUK is a severe inflammatory disorder of the cornea characterized by progressive crescent-shaped thinning of the peripheral stromal layer, with or without an overlying epithelial defect, dense inflammatory infiltration, and edema. Conditions leading to PUK include systemic infectious diseases, non-infectious autoimmune or inflammatory disorders, and primary ocular issues.12,123
Epidemiologic featuresThe incidence of PUK ranges from 0.2 cases per 3 million population per year.131 Collagen vascular diseases and small-vessel granulomatous vasculitides coexist in approximately 50% of PUK cases. Although PUK occurs in only 2% of RA patients, it is the most common rheumatic disease associated with RA (33–66%). Typically, RA-associated PUK affects women in their fifth or sixth decade of life who have a longstanding diagnosis of RA.12 However, PUK, as the initial manifestation of RA without joint involvement, has been reported.132
PathogenesisThe peripheral cornea is a crucial transition zone where the anatomical and functional characteristics of the conjunctiva, sclera, and cornea intersect. Consequently, the corneal periphery drains into conjunctival lymphatic and blood vessels. Compared to the central and paracentral zones, the corneal periphery is thicker and less sensitive, exhibits a flatter curvature, and has a higher density of C1, IgM, LCs, and inflammatory cells. The transition from sclera to cornea and from conjunctival to corneal epithelium features unique niches primarily occupied by stem cells with a very high replication and transformation rate. These differences make the corneal periphery vulnerable to hypersensitivity and immunologic reactions.133
In contrast, the extracellular matrix (ECM), which consists of layers of collagen fibrils embedded in a framework of glycosaminoglycans, is highly organized within the central and paracentral cornea compared to the irregular structures found in the periphery.131 The underlying corneal fibroblasts (keratocytes) maintain the integrity of the ECM within these lamellae. In contrast, the delicate balance between collagenases and their specific tissue inhibitors regulates the turnover rate of the corneal matrix.134 An imbalance between MMP-1, an ECM-degrading interstitial collagenase, and its tissue inhibitor of MMP (TIMP)-1, which facilitates keratolysis, is considered the primary pathogenic mechanism behind PUK. Local fibroblasts, macrophages, and granulocytes produce MMP-1.134,135 While collagenases are expressed in the stromal tissue of ulcerative RA corneas, they are absent in corneal donor buttons of patients without RA.135
On the other hand, the levels of TIMP-1 found in RA corneas were significantly lower than those in the controls. Other collagenases, such as MMP-2 and MMP-9, hydrolyze type IV collagen, a basement membrane component. While corneal keratocytes produce MMP-2, the source of MMP-9 remains unclear. Smith et al. observed increased MMP-9 secretion in tears from patients with RA-associated PUK compared to healthy controls, indicating that MMP-9 could be secreted by the lacrimal gland, corneal and conjunctival epithelium, or by the inflammatory cells (neutrophils and macrophages) that accumulate in the vascular limbus of PUK eyes.136
Galor and Thorne proposed that PUK may arise from the abnormal activation of T cells, resulting in antibody production and the deposition of immune complexes, reflecting a type III hypersensitivity reaction.137 In RA patients, self-antigens and autoantibodies also form immune complexes, further activating complement proteins and B-cells, which may produce more autoantibodies and cytokines. This may trigger abnormal T-cell responses, leading to complement activation via C1 that induces recruitment and stromal infiltration of inflammatory cells.131,133
Clinical manifestations and diagnosisPUK patients typically present with an acute onset of tearing, injection, photophobia, foreign body sensation, and pain of variable intensity with or without vision loss. Slit-lamp examination reveals a crescent-shaped peripheral ulcer with stromal thinning and infiltration (Fig. 5).131 A varying degree of inflammation involving the adjacent conjunctiva and sclera can also be seen.137 The most extensive series of RA-associated PUK eyes reported concomitant anterior diffuse, nodular, and necrotizing scleritis in 46%, 21%, and 15% of eyes at PUK onset, respectively.12

Corneal ulceration associated with Rheumatoid Arthritis (RA). (A) Peripheral ulcerative keratitis in an RA patient with associated Sjögren syndrome, showing a dense inferior peripheral circumferential stromal infiltrate. (B) Positive fluorescein staining of the same eye showing an extensive epithelial defect. (C) Central corneal ulceration (positive fluorescein staining) in an aged woman with a long history of uncontrolled RA.
Of most significant importance, Foster et al. reported a 50% mortality rate at ten years in RA patients presenting with concomitant necrotizing scleritis and PUK not managed with IMT.14 These results were later supported by Ogra et al., who found an increased risk of death between patients with PUK, necrotizing scleritis, or both, between those receiving IMT and those without. The authors estimated a mean time to death of 10.7 years in patients who did not receive IMT and 24.7 years in those who did receive IMT.123 Ruiz-Lozano et al. evaluated the role of early IMT, defined as treatment administration within the first four weeks from PUK onset in 36 patients (52 eyes) with RA-associated PUK.12 The authors reported that early IMT conferred protection for PUK recurrence, whereas late IMT and an unknown RA presence at the PUK onset were associated with significant vision loss.12 For those with concomitant scleritis, the need for patient-tailored treatment is emphasized, as it is recommended over protocol-guided approaches.138
ComplicationsCorneal perforation is a serious and feared acute complication, primarily observed in patients with sSS, multiple recurrences, poor treatment compliance, or late or inadequate immunosuppression. Chronic and recurrent PUK can result in irregular corneal astigmatism, opacity, scarring, and decreased vision.131 A retrospective case series that examined 52 eyes affected by PUK, with an average follow-up of 7 years, found that 19% of the eyes experienced moderate visual loss, while 12% experienced severe visual loss. Four patients developed corneal perforations, with two of these patients having repeated perforations in both eyes. Additional complications reported included cataracts (10 cases), elevated intraocular pressure (5 cases), and glaucoma (3 cases).123
Central and paracentral corneal ulceration (CPCU)Definition and backgroundCPCU, a distinct clinical entity that can develop in the corneas of patients with RA with or without associated sSS, even if the disease is unknown or inactive at the onset.139 This corneal ulceration, distinct from PUK, is characterized by its subtle or absent symptoms, mild or unnoticed ocular surface inflammation, and central or paracentral location. These rapidly settled sterile corneal melts pose a treatment challenge and often recur, potentially leading to perforation and necessitating urgent tectonic grafting procedures. While it is not a common occurrence, the rapidly progressive and smoldering clinical course of CPCU underscores the need for immediate recognition. The prompt initiation of aggressive IMT and supportive topical treatment is crucial to prevent potential ocular integrity loss and severe visual consequences.139,140
Epidemiologic featuresIn a study of 589 patients with RA, Watanabe et al. reported that 1.4% developed corneal ulceration.141 In a UK hospital case series on rheumatoid corneal ulcers, 40 eyes were documented with keratolysis. Among these, 10 ulcers (25%) were identified as paracentral, while 7 ulcers (17%) were central. Notably, only 7 eyes (18%) showed elevated inflammatory markers.142 The remaining literature on this topic consists primarily of case reports.139,143,144
PathogenesisThe mechanisms involved in CPCU have not been thoroughly elucidated. However, there are theories suggesting that immune-mediated processes and structural anomalies in collagen may play a role. Evidence supporting the role of the adaptive immune response includes the presence of immune cells, the overexpression of inflammatory mediators, and positive responses to anti-inflammatory treatments.139,145,146
Immunohistopathologic analysis of two frozen corneal buttons from CPCU cases demonstrated epithelial and stromal abnormalities.145 Epithelial IgG and IgM deposits and HLA-DR expression in epithelial cells and stromal keratocytes have been detected. However, inflammatory cells, including macrophages, T-lymphocytes, and neutrophils, were abundant in the ulcer margins, suggesting a complex adaptative immune response with significant cellular participation. Moreover, IL-6 and TNF-α have been detected in corneal buttons of patients who developed RA-related ulcerations or perforations.146 It is noteworthy to mention that IL-1 and TNF-α are inflammatory mediators found in the synovial fluid of patients with RA.145 IL-6 regulates collagen synthesis and MMP production in keratocytes, while TNF is involved in various pathologic processes across different cell types. On the other hand, IL-1 activates protease and collagenase production in synovial fibroblasts. It is hypothesized that IL-1 may also induce collagenase activation in corneal stromal keratocytes.145
The imbalance between collagen synthesis and degradation has been further supported by finding a reduced TIMP-1 expression in ulcerated RA corneas.135 Such pathogenic features lead to increased levels of collagenase activity and macrophage presence in the epithelium that mediate tissue destruction.
Risk factors facilitating the appearance and progression of CPCU include KCS- and sSS-associated ocular surface dryness and local trauma due to the clear-cornea cataract phacoemulsification surgery technique.142 It is hypothesized that apart from corneal stromal incisional and thermic trauma and sensory nerves section, anterior segment surgical interventions impact the already deteriorated ocular surface of KCS patients, predisposing them to CPCU.147,148
Topical medications’ toxicity and potential corneal dysfunction modifications may also be involved in the development of CPCU. Corticosteroids can impair the normal wound-healing processes by reducing collagen synthesis.149 Generic NSAIDs, commonly used to prevent cystoid macular edema after cataract surgery, have also been related to corneal melts, although this is a rare side effect. After cataract extraction, these agents can produce mild anesthesia that reduces corneal sensitivity, resulting in reduced wound healing.143
Clinical manifestations and diagnosisCPCU typically presents with a painless, sterile, non-infiltrated central or paracentral corneal ulceration that can develop in patients with unknown RA or during a quiescence stage (Fig. 5).139 This acute onset, rapidly progressive ulceration is classically characterized by a low-inflammatory framework marked by faint or absent stromal infiltration, ciliary injection, or limbal vasculitis. In cases with RA history, the ulcers mainly developed in patients with longstanding disease (10–20 years) or those with a severe or uncontrolled persistent inflammatory clinical course. If the patient is undiagnosed, it is essential to establish the diagnosis by a complete rheumatologic examination and performing a pertinent imaging and laboratory workup, including the rheumatoid factor, anti-CCP, and antinuclear antibody titers.150
TreatmentManagement of CPCU can prove challenging, as the ulceration commonly progresses rapidly to stromal melting and even perforation, giving little or no time for medical intervention. In the case of descemetocele or perforation, tissue adhesive to seal the defect as a “bridge” for keratoplasty can be performed as a temporary therapeutic remedy. Kervick et al. managed five patients (seven eyes) with systemic immunosuppression and tissue adhesive with a BCL or tectonic keratoplasty.145 However, all these ulcers recurred and required re-grafting. In 5 eyes with recurrent ulceration, re-epithelization, and closure of the defects were attained after introducing topical cyclosporine 2%, suggesting that it may be an effective initial treatment. Patch grafts and tissue adhesives proved helpful in one report.144 One case of a patient who declined keratoplasty was successfully treated with a Gundersen conjunctival flap.150
ComplicationsIt is not uncommon for CPCU to progress from a partial-thickness corneal ulcer to a large perforation.139 In these cases, a shallow anterior chamber and prolapsed iris can be seen upon slit lamp examination. These patients will need emergency surgical management, a procedure that carries the risk of the graft being placed eccentrically, inducing irregular astigmatism and decreased vision.143 Unfortunately, these ulcers can also recur in the grafts and cause them to fail.140
ConclusionsAny signs of ocular inflammation, including red eye, pain, photophobia, and blurred vision, in patients with RA can lead to significant morbidity if not recognized promptly or treated adequately. Rheumatologists should be vigilant for these signs and symptoms and should be aware of the wide range of ocular manifestations in RA patients, as they often indicate poorly controlled systemic disease. It is also important to recognize that these ocular inflammatory manifestations can not only reflect a reactivation of systemic disease but can also lead to considerable complications, especially in cases of RA with a new onset of necrotizing scleritis and peripheral ulcerative keratitis.
Conversely, eye care specialists should suspect an underlying active systemic autoimmune disease in patients with unresponsive KCS, episcleritis, scleritis, PUK, and CPCU, particularly since ocular symptoms may be the initial manifestation of RA. In such cases, a thorough ocular and systemic serologic screening should be conducted, followed by an immediate referral to a rheumatologist for further evaluation and treatment.
Ultimately, the anatomical and visual outcomes depend on individual expertise and effective multidisciplinary communication and management between ophthalmologists and rheumatologists. This collaboration is essential for achieving comprehensive systemic disease control through effective combinations of corticosteroids, immunosuppressive agents, and biologic therapies.
Methods of literature searchThe authors conducted an extensive literature search using the National Library of Medicine's PubMed, Embase, Web of Science, and Google Scholar database for all English language articles published until July 2024. The following search string was used: (“rheumatoid arthritis”) AND (“dry eye disease” OR “dry eye” OR “episcleritis” OR “scleritis” OR “peripheral ulcerative keratitis” OR “central corneal ulcer*” OR “paracentral corneal ulcer*” OR “ocular manifestations” OR “ocular involvement”). Case reports, case series, letters to the editor, review articles, and original articles were included. Relevant references within articles found were also included. Articles in languages other than English were excluded.
FundingNone.
Conflict of interestNone.
The authors would like to thank Dr. Jesus Alberto Cardenas de la Garza for his advice during the development of this review.


