



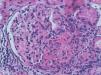
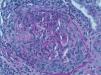
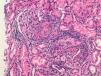
The development of vasculitis in patients with systemic sclerosis (SS) is a rare event and association. However, because they share similar clinical manifestations but a different etiopathogenic basis, the need to distinguish between inflammatory vasculitic damage or scleroderma vasculopathy can sometimes be a real dilemma that determines the clinical prognosis and therapeutic regimen for these patients.
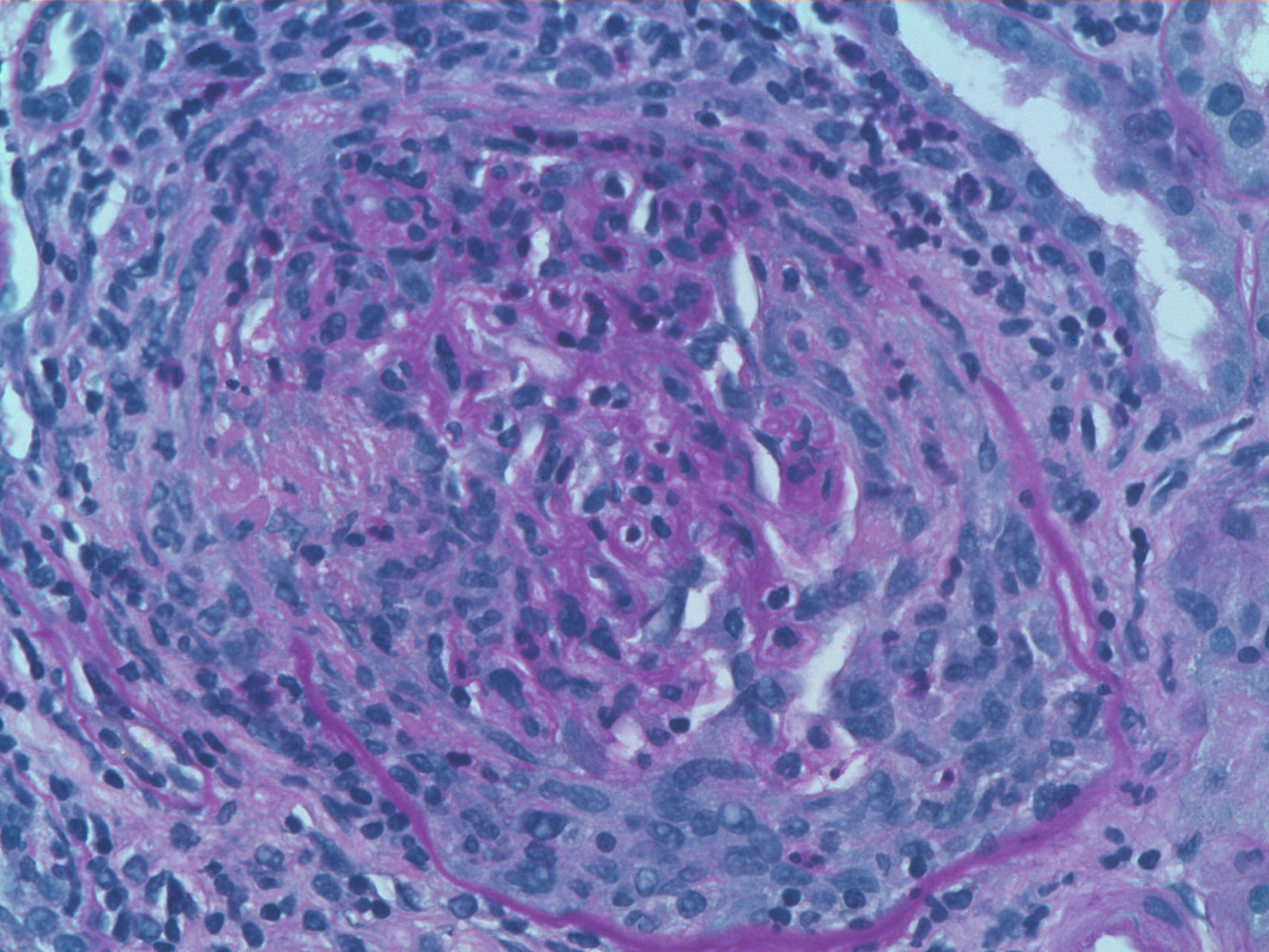
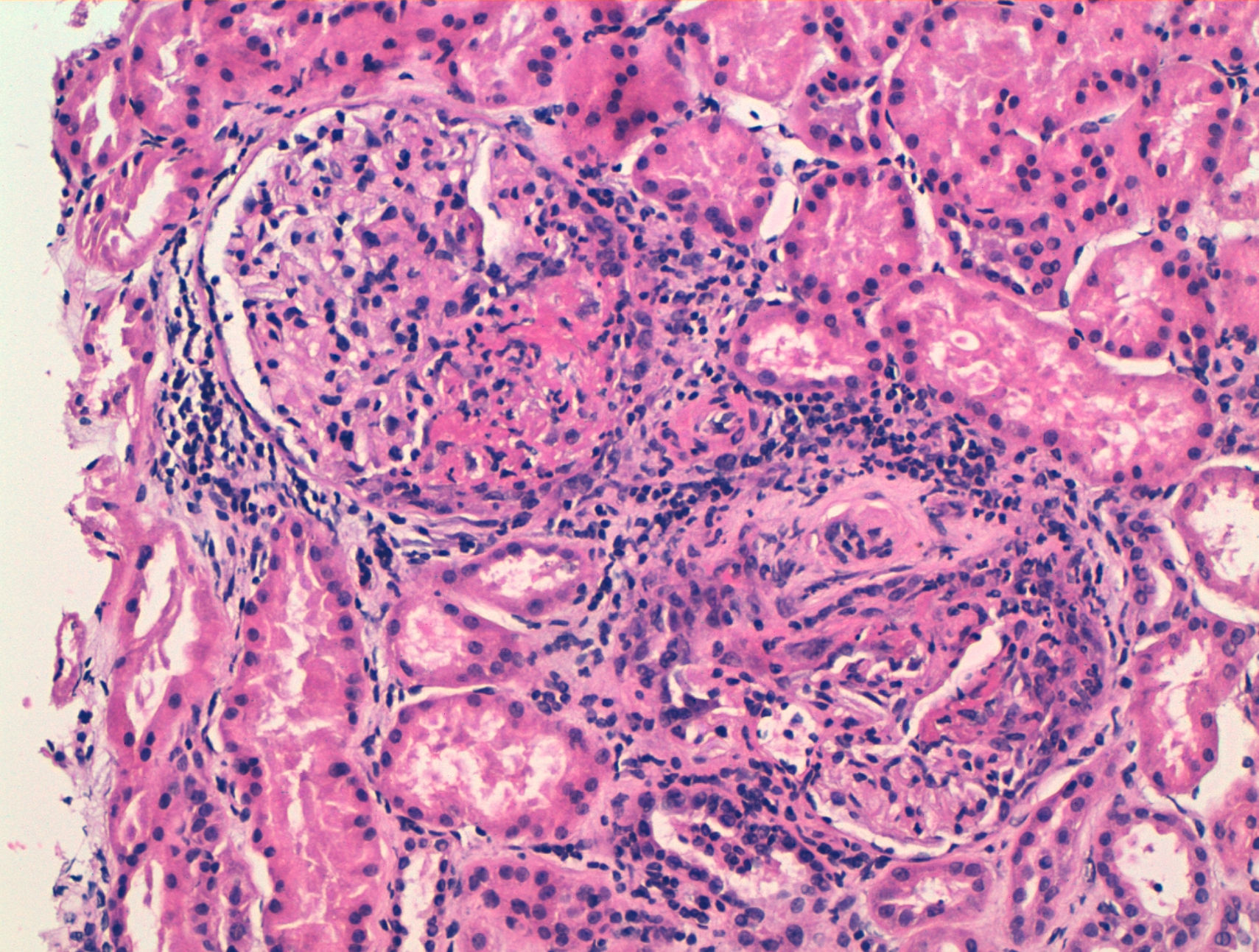
We report the case of a 63-year-old woman with a history of hypertension treated with inhibitors of the angiotensin converting enzyme (ACE) inhibitors and dyslipidemia. She was diagnosed 16 years before with limited cutaneous SS based on Raynaud's phenomenon, sclerodactyly, telangiectasias, esophageal dysfunction, and mild pulmonary fibrosis of both lung bases, with no functional impact on blood gases, with negative anticentromere, Jo1, Scl-70, and PM-1 antibodies. She had not received specific treatment except for oral corticosteroids for an acute episode of pleuropericarditis, presenting a secondary cardiac arrest one year prior to admission. She came to the clinic due to fatigue and malaise of a month's duration, without fever and no other data of interest. Physical examination showed BP 150/90mmHg and typical stigmas of scleroderma, mucocutaneous pallor, small laterocervical lymphadenopathy, fine crackles, and mild edema of the lower extremities, without evidence of heart failure. Laboratory tests showed anemia with hemoglobin of 10.8g/dl, (creatinine of 2.2mg/dl, with no known impairment of renal function, having been previously normal) CRP of 48mg/dl, and an ESR of 58mm/h. Immunological tests showed positive ANA 1/160 with a nucleolar pattern, ENA (Ro, La, RNP, Sm) and negative anti-GBM antibody, normal C3 and C4 and positive 1/640ANCA p-ANCA, with positive anti-MPO ELISA 28U/ml (negative 0–7) and negative anti-PR3. The urinalysis showed microhematuria and mild proteinuria of 21mg/dl (0.6g/d). The chest X-ray was normal, except for mild fibrosis in both lung bases. We performed a kidney biopsy. We appreciated 18 glomeruli, 6 of them completely sclerotic. Of the remaining 12, 7 had severe extracapillary proliferation forming circumferential crescents (Figs. 1 and 2) that engulfed the glomerular tuft, and in 5 of these glomeruli were showed images of vascular plume fibrinoid necrosis (Fig. 3). The rest of the glomeruli showed no lesions. Direct immunofluorescence was negative for IgG, IgA, IgM, and C3. In the interstitium there was marked inflammatory infiltrates and vasculitis lesions were visible, especially in small and medium caliber vessels. This supported pauci-immune extracapillary glomerulonephritis. Treatment was initiated with 3 induction boluses of methylprednisolone 1g/day and thereafter iv prednisone at 1mg/kg/day orally with subsequent reduction, along with monthly cyclophosphamide iv boluses at a dose of 0.5mg/m2. The initial response was poor, with deterioration of the renal function reaching creatinine levels of 5.4mg/dl. At 6 months, after 5 cycles of cyclophosphamide, creatinine stabilized at 2mg/dl and negative ANCA, so we continued treatment with azathioprine 100mg/day. At 2 months it had to be replaced due to moderate hepatotoxicity, using MMF 500mg/12h, finally suspendedshe has not undergone any more hospitalizations. However, residual impairment of renal function with creatinine of 2mg/dl persists.
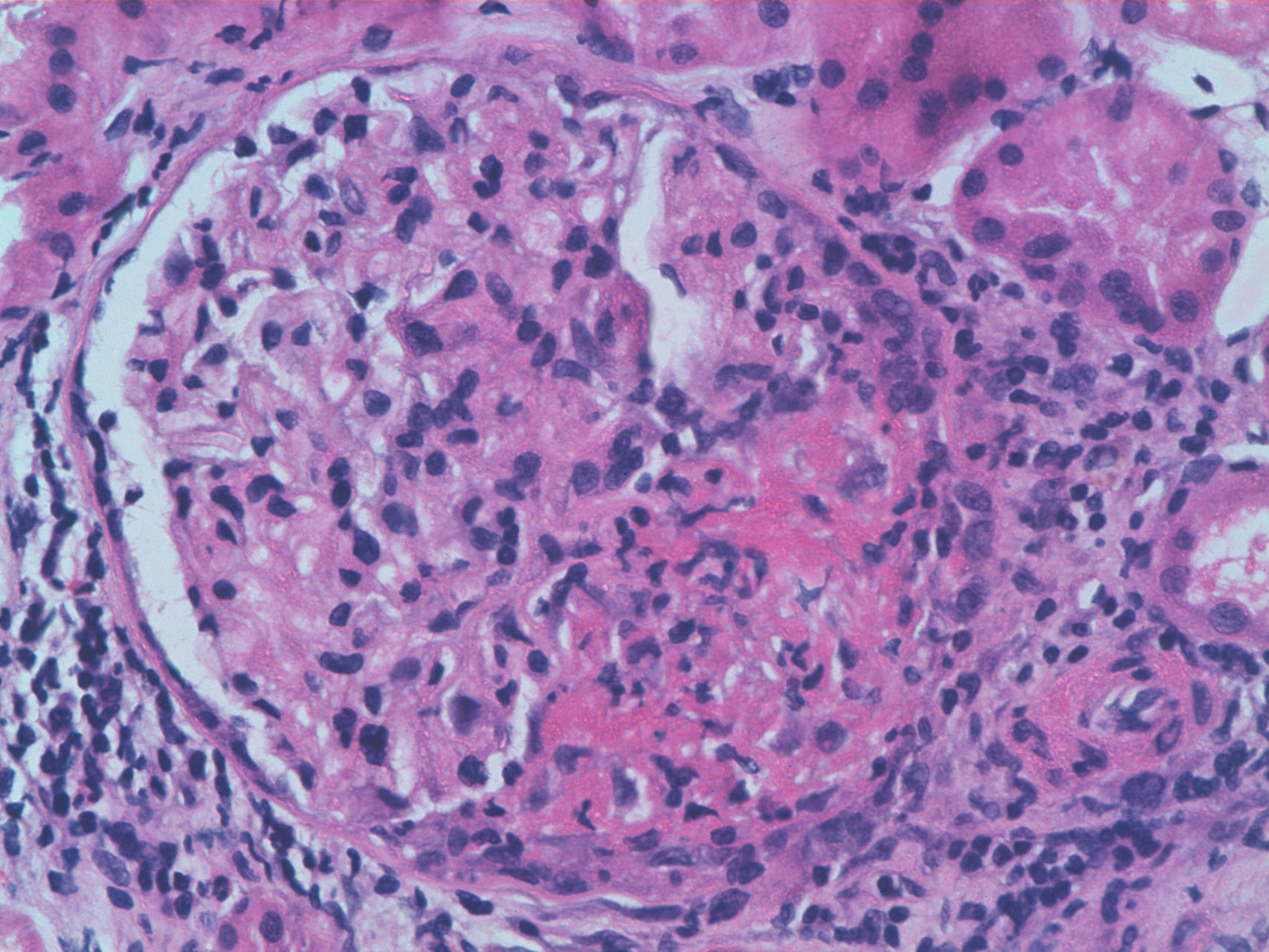
Renal involvement in SS is characterized by obliterative vascular disease of the cortical arteries due to endothelial damage, vascular wall thickening, and renal hypoperfusion juxtaglomerular hyperplasia, leading to a state of hyperreninemia. The most serious clinical consequences, the scleroderma crisis (SC), appears in 10%–15% of patients with SS, being more frequent in diffuse forms and after the 3 or 4 first years from disease onset. Some risk factors have been associated as risk factors, such as anemia, the use of corticosteroids (>15mg prednisone), black ethnicity, and topoisomerase III antibodies, but not anticentromere or anti-Scl70 antibodies. It can classically be accompanied by malignant hypertension, acute kidney injury, and microangiopathic hemolytic anemia. Although the prognosis has improved with treatment with ACE inhibitors, estimated mortality is still approximately 19% per year and 40% at 5 years.1
However, our case presented an atypical nephropathy in the biopsy. ANCA associated vasculitis (AAV) is the most frequent type of vasculitis seen in SS, with large and medium caliber vasculitis being even more exceptional.2 The incidence of ANCA in patients with SS varies between 7% and 13%, but clinical impact is rareç; unknown pathophysiological mechanism trigger the development of vasculitis, even on previous sclerotic lesions. Patients with MPO-ANCA vasculitis are more susceptible to present clinical features and PR3-ANCA rarely have clinical consequences, with involvement of upper airway or granulomatous lesions being more common. Rho et al., in a review of 50 AAV cases associated to SS3 describe female predominance (84%), with a mean age of 57 years and a mean duration of SS of 9.5 years before the onset of vasculitis. It has been related to the previous use of d-penicillamine. Renal involvement is the most frequent (82%), followed by gastrointestinal (58%) and alveolar hemorrhage (22%). Only 28% present hypertension. Distal ischemia or cutaneous vasculitis are less common manifestations. No differences were found between diffuse and limited forms, with anti-Scl70 being clearly prevalent (70%) compared to anti-centromere antibodies (14%). Seventy-two percent of cases are associated with anti-MPO compared with 24% of anti-PR-3 positive patients, most with a p-ANCA pattern by IF. High titers of p-ANCA appear to correlate with a rapid deterioration of renal function.4 Arad et al. added prognostic information such as improvement in 51% of cases and evolution to ESRD or death in 14% and 34%, respectively. Mortality occurs mostly during the first year from the onset of vasculitis, mainly due to secondary causes as infectious disease and pulmonary hemorrhage. Mortality contrasts with the SC, which is mainly caused by late complications of chronic dialysis.5
Success in treating the etiopathogenesis is less of a focusthan avoiding the likely irreversible damage. Suggestive manifestations for differential diagnosis are SC hypertension (88%), thrombocytopenia and microangiopathic hemolytic anemia (50%), typically appearing early in the SC and with rare presentations of elevation of the RFA or fever. If associated to pulmonary vasculitis, severe worsening of anemia without evidence of hemolysis with elevated RFA may be key to the interpretation of a radiological pattern of alveolar hemorrhage on pulmonary fibrosis or cardiac edema.
There are no specific recommendations for AAV treatment associated to SS. The common trend is to follow the standard recommendations of AAV treatment with steroids and cyclophosphamide boluses. High-dose corticosteroid therapy has proven effective and renal crisis did not appear in the case described. Plasma exchange has also been used, and more recently rituximab, with favorable6 results.
The association of AAV in patients with SS is uncommon. The most common clinical presentation is often long standing renal p-ANCA in positive Scl70 patients. It constitutes a differential diagnostic challenge with a scleroderma renal crisis and especially in unusual normotensive presentations. Immunosuppressive therapy is effective, but with significant mortality or progression to ESRD.
Please, cite this article as: Zurita Prada PA, et al. Vasculitis ANCA positivo en un paciente con esclerosis sistémica. Reumatol Clin. 2013;9:72–74.


