




La primera publicación moderna sobre esclerodermia se atribuye generalmente al italiano Curzio en 1753. Más tarde, Alibert afirmó ser el primero en reconocer la enfermedad, la llamó“sclermia circumscripia”, y describió 2 casos de lo que fue probablemente la esclerodermia lineal1,2.
La esclerodermia puede clasificarse en sistémica, localizada y síndromes que semejan esclerodermia. Es una enfermedad autoinmunitaria de etiología desconocida y caracterizada por fibrosis.
La esclerodermia sistémica (ES) afecta órganos como piel, aparato digestivo, pulmones, corazón y riñones. Recientemente, un comité de criterios de clasificación para esclerodermia sistémica juvenil que incluía miembros de la Sociedad Europea de Reumatología Pediátrica, el Colegio Americano de Reumatología y la Liga Europea Contra Reumatismo desarrolló nuevos criterios de clasificación para ayudar a normalizar la realización de investigaciones en esta rara enfermedad pediátrica3,4.
Comúnmente, la esclerodermia localizada (EL) se ha dividido en 3 grupos: morfea, morfea generalizada y esclerodermia lineal. En el año 2004, la Sociedad Europea de Reumatología Pediátrica propuso nuevos criterios de clasificación para EL juveniles e incluyeron 5 subtipos: morfea circunscripta, esclerodermia lineal, morfea generalizada, morfea panesclerótica y esclerodermia mixta (cuando se presenta una combinación de 2 o más de los subtipos anteriores)5.
El inicio de la esclerodermia en niños es raro con menos del 5% de los casos en menores de 16 años, con una edad media de 8,1 años y con un pico entre los 10 y los 16 años de edad, según la literatura médica mundial5–7.
La esclerodermia y sus diferentes subtipos presentan variabilidad en su prevalencia en diferentes regiones del mundo, por lo que factores genéticos, raciales y ambientales parecen estar involucrados. Con el presente estudio se pretende describir las características clínicas y de laboratorio de un grupo de 62 pacientes en edad pediátrica con esclerodermia en un centro de tercer nivel.
Materiales y métodosSe revisaron las historias clínicas de los niños con diagnóstico de esclerodermia en el Hospital Infantil de México Federico Gómez (centro de referencia de tercer nivel) entre 2000 y 2007. El diagnóstico se realizó según datos clínicos, biopsia de piel, participación de un órgano interno, anomalías vasculares (incluido el fenómeno de Raynaud) y presencia de autoanticuerpos, como anticuerpos antinucleares (ANA), anti-Scl70 y anticentrómero.
En el presente estudio se aplicaron los nuevos criterios de clasificación preliminar para ES. Asimismo, se excluyó a los pacientes con otras enfermedades autoinmunitarias.
Se registraron los siguientes datos al inicio y durante el seguimiento de los pacientes: sexo, edad al inicio de la enfermedad y al diagnóstico, consultas previas, estado nutricional, presencia de alteraciones cutáneas (edema, esclerosis, esclerodactilia, calcinosis, punteados digitales, telangiectasias y engrosamiento de la piel), alteraciones cardiovasculares (fenómeno de Raynaud, pericarditis, hipertensión, insuficiencia cardíaca, arritmias e hipertensión pulmonar), alteraciones respiratorias (tos, pleuritis, fibrosis, alteraciones en espirometría y pletismografía), alteraciones gastrointestinales (disfagia, diarrea, estreñimiento y reflujo gastroesofágico [RGE]), alteraciones musculoesqueléticas (artritis y afección muscular), alteraciones renales (proteinuria) y otras (pérdida de peso, retraso del crecimiento y crisis convulsivas). Además, se registró la presencia de autoanticuerpos, como ANA, anti-RNP, anti-Sm, anti-Scl70 y anti-centrómero, y el tratamiento recibido (corticoide, inmunosupresores y vasodilatadores).
ResultadosCaracterísticas generalesSe incluyó a 62 pacientes, entre 2000 y 2007, con diagnóstico de esclerodermia; 21 pacientes (34%) de sexo masculino y 41 pacientes (66%) de sexo femenino. La relación mujer/varón fue de 2:1. El diagnóstico de esclerodermia fue más frecuente en el grupo comprendido entre 5 y 11 años de edad con 39 pacientes (63%), con una edad media de 7,8 años (rango de 1 a 14).
Consultas previas al ingreso: es importante destacar que el 100% de los pacientes consultó previamente con personal médico antes del diagnóstico y, a su vez, 54 pacientes (87%) consultaron más de 2 veces. En cuanto a la consulta con personal no médico, 5 pacientes (8%) la realizaron en una ocasión y 13 pacientes (21%) en 2 o más ocasiones.
En cuanto al tiempo de evolución clínica previo al diagnóstico, el 60% de los pacientes presentó lesiones en la piel sin diagnóstico por uno o más años y el 40% de los pacientes estuvo sin diagnóstico por meses. De estos últimos, sólo 3 pacientes se diagnosticaron al mes de iniciadas las lesiones. Al momento del diagnóstico, la duración de la enfermedad, en general, fue de una media de 1,9 años (rango de un mes a 10 años). En tanto que en la ES tuvo una media de 2,4 años (rango de un mes a 10 años) y en la EL una media de 1,6 años (rango de un mes a 16,7 años).
Antecedentes previos a la enfermedad: del total de pacientes, 11 (18%) presentaron antecedentes previos a la enfermedad, de los que sólo 4 pacientes revelaron enfermedades infecciosas respiratorias previas al cuadro, 3 pacientes tenían historia de malformaciones y los restantes 4 pacientes refirieron traumatismos previos (tabla 1).
Tabla 1. Características demográficas principales de 62 pacientes con esclerodermia
| Todos | Esclerodermia sistémica | Esclerodermia localizada | |
| Pacientes | 62 | 18 | 44 |
| Sexo (mujer/varón) | 2/1 | 2,6/1 | 1,75/1 |
| Varones | 21 | 5 | 16 |
| Mujeres | 41 | 13 | 28 |
| Edad de inicio de síntomas (año/mes) | |||
| Media | 7/10 | 7/8 | 7/11 |
| Mediana | 7/0 | 7/6 | 6/6 |
| Rango | 1–14 años | 2–14 años | 1–14 años |
| Duración de la enfermedad al diagnóstico (año/mes) | |||
| Media | 1/11 | 3/2 | 0/7 |
| Mediana | 1/0 | 2/0 | 1/0 |
| Rango | 1 mes–10 años | 1 mes–10 años | 1 mes–10 años |
| Factores ambientales | 8 | 2 | 6 |
Estado nutricional: la mayoría de los pacientes eran eutróficos, sumando un total de 51 pacientes (82%). Aun así, hubo un número importante de desnutrición: 11 pacientes (18%), de los que 6 (10%) presentaban riesgo nutricional, 4 (6%) presentaban desnutrición moderada y uno (2%) presentaba desnutrición grave con aspecto caquéctico.
Características clínicasSe encontraron 18 casos (29%) con ES y 44 casos (71%) con EL. Del total, a su vez, 7 pacientes (11%) tuvieron esclerodermia lineal de tipo Parry-Romberg, 3 pacientes (4,5%) con esclerodermia lineal de tipo golpe de sable y 6 pacientes con ambas lesiones a la vez. (figs. 1 y 2)
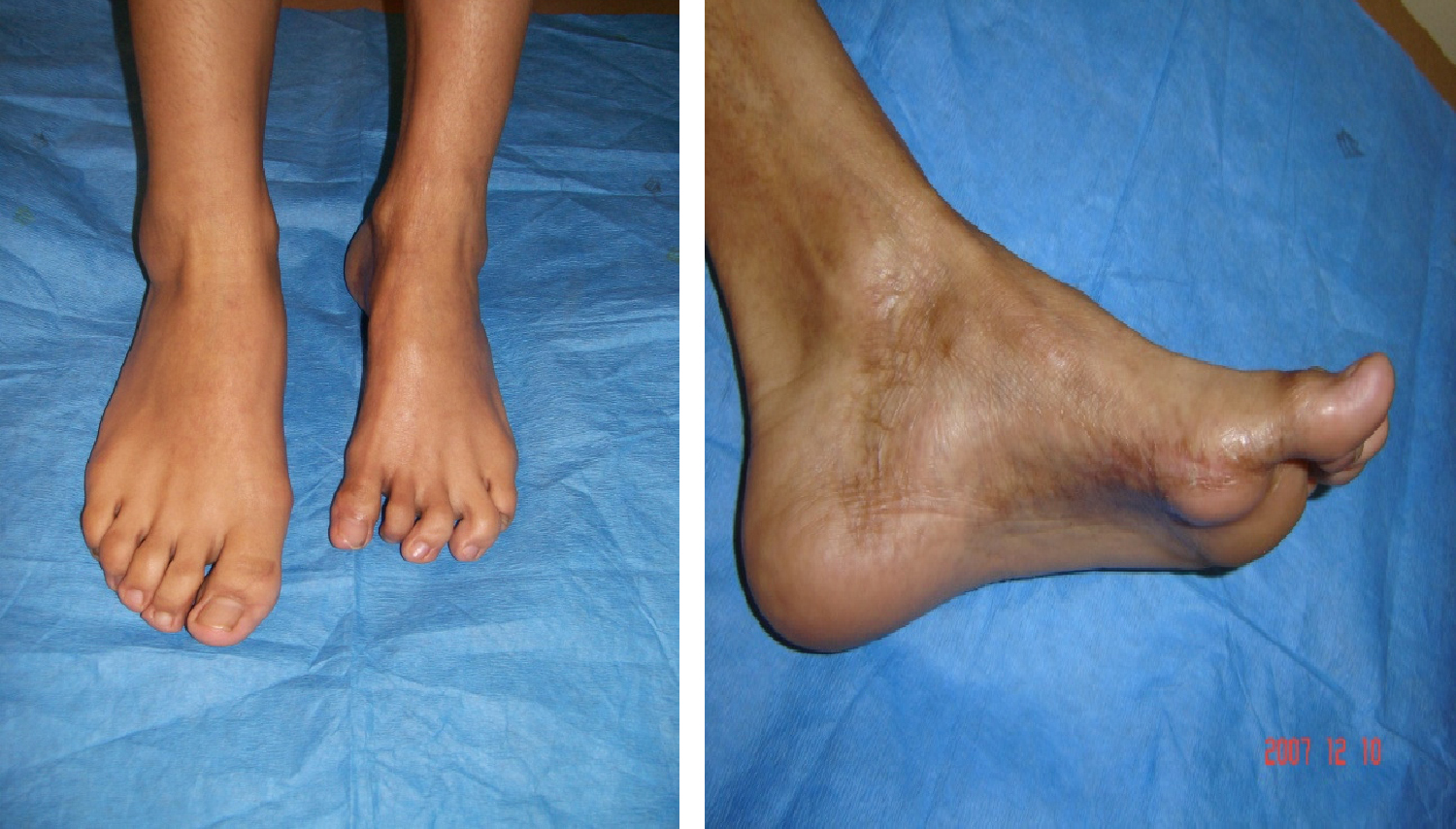
Figura 1. Paciente con esclerodermia sistémica y lesión extensa. Fuente: pacientes de la Clínica de Enfermedades por Daño Inmunológico.
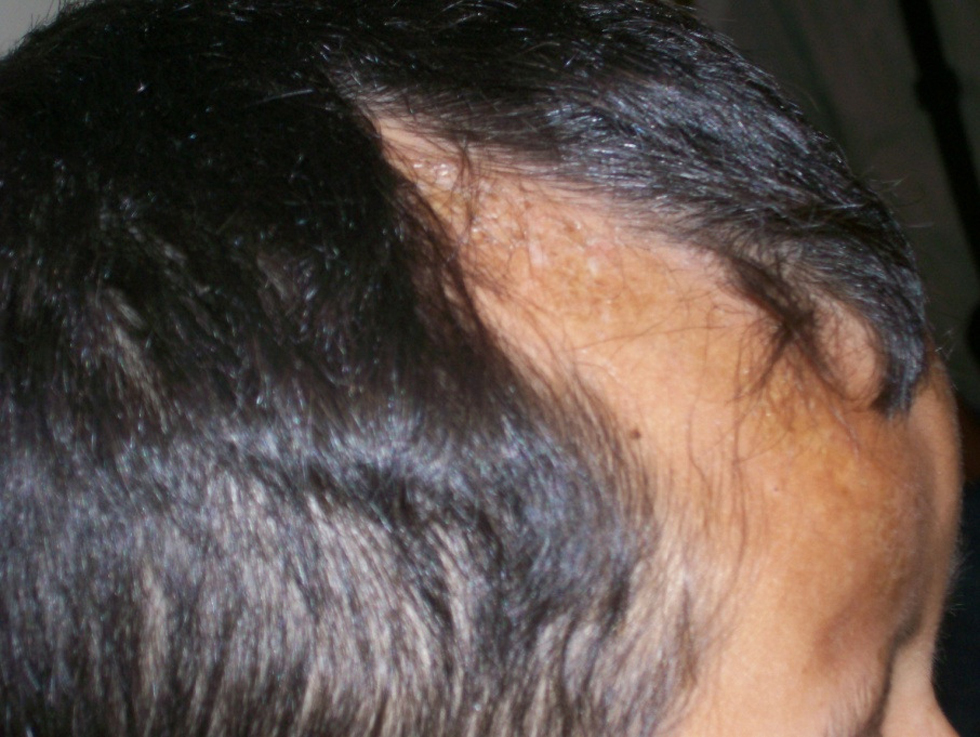
Figura 2. Paciente con esclerodermia lineal de tipo golpe de sable en región frontoparietal derecha. Fuente: pacientes de la Clínica de Enfermedades por Daño Inmunológico.
El comienzo de las lesiones fue insidioso en todos los pacientes. Entre los síntomas y los signos asociados en pacientes con ES, se pueden mencionar astenia, hiporexia, pirosis, dolor en epigastrio, tos, artralgias, livedo reticularis, telangiectasias, calcinosis y fenómeno de Raynaud. Once pacientes (61%) presentaron disfagia, pirosis y dolor en epigastrio; sólo 2 pacientes presentaron tos en el transcurso de su seguimiento; 10 pacientes (56%) presentaron calcinosis (fig. 3); 16 pacientes (89%) presentaron fenómeno de Raynaud, 14 lo presentaron en dedos de manos y 2 en dedos de manos y pies; 5 pacientes (28%) presentaron artralgias, solamente uno de ellos presentó artritis oligoarticular pasajera, y 11 pacientes (61%) presentaron livedo reticularis y telangiectasias. También se observaron convulsiones en 3 pacientes, 2 de ellos con esclerodermia lineal de tipo golpe de sable. En algunos casos, también se encontró debilidad muscular, edema y en un caso, afección renal. No se encontraron complicaciones cardíacas, endocrinológicas ni oculares (tabla 2).
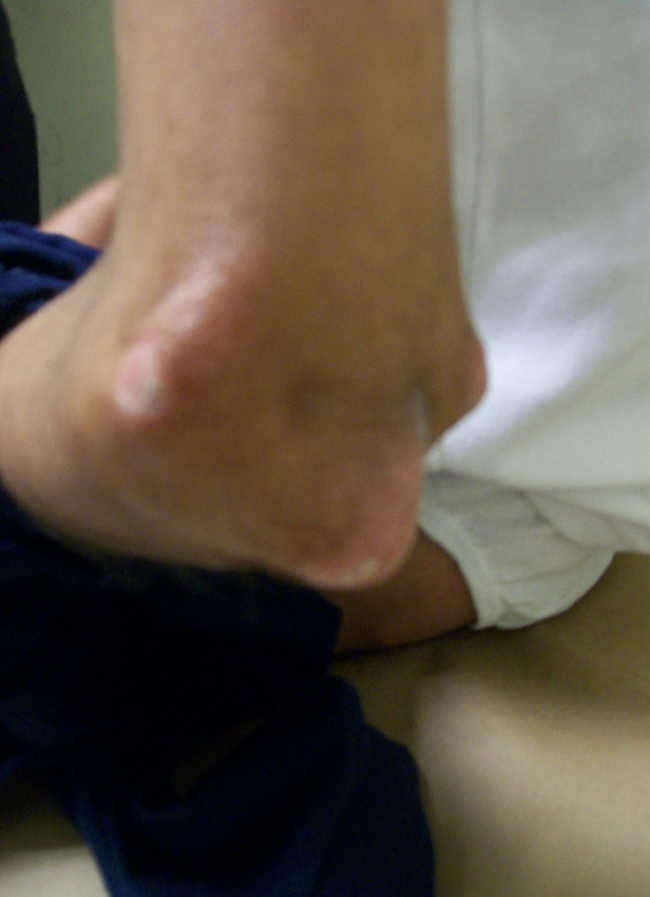
Figura 3. Calcinosis en codo izquierdo en un paciente con esclerodermia sistémica. Fuente: pacientes de la Clínica de Enfermedades por Daño Inmunológico.
Tabla 2. Frecuencia de signos y síntomas en esclerodermia sistémica (n = 18)
| Síntoma | Pacientes, n | (%) |
| Esclerosis proximal | 16 | 89 |
| Esclerodactilia | 12 | 67 |
| Fenómeno de Raynaud | 16 | 89 |
| Artralgias | 4 | 22 |
| Artritis | 1 | 6 |
| Edema | 6 | 33 |
| Calcinosis | 10 | 56 |
| Disfagia, pirosis y dolor en epigastrio | 11 | 61 |
| Debilidad muscular | 2 | 11 |
| Tos | 2 | 11 |
| Livedo reticularis | 11 | 61 |
En cuanto a los tipos de morfea, en 16 pacientes (26%) se observó EL facial, 10 pacientes (16%) presentaron EL extrafacial, en 12 pacientes (19%) fue morfea circunscripta, en 2 pacientes (4%) morfea panesclerótica, en 8 pacientes (13%) morfea generalizada y en 14 pacientes (22%) morfea mixta.
Variables de estudiosRespecto a los estudios de laboratorio, en la biometría hemática 5 pacientes (8%) presentaron leucocitopenia, de los que 2 tuvieron linfocitopenia. Se observó anemia en 10 pacientes de tipo normocrómica, normocítica. En cuanto a los reactantes de fase aguda, 11 pacientes (18%) presentaron velocidad de sedimentación globular acelerada y la reacción en cadena de la polimerasa PCR se realizó en 12 pacientes (19%), encontrándose elevada en 4 de ellos (6%).
Se realizó análisis de orina en todos los casos, con resultado patológico (proteinuria) sólo en el paciente que ameritó biopsia renal, encontrándose glomeruloesclerosis segmentaria y lesiones vasculares en arteriolas, compatible con esclerodermia. En cuanto a la funcionalidad renal, todos los pacientes presentaron valores normales de urea y de creatinina. Tres pacientes presentaron alteraciones en las aminotransferasas, con elevación de éstas durante el tratamiento, lo que ameritó cambio de tratamiento.
Dentro de las pruebas inmunológicas se determinaron los siguientes anticuerpos: ANA en 35 pacientes (56%), con 14 casos positivos (10 para ES y 4 para EL); anti-Scl70 en 19 pacientes (31%), con sólo 2 casos positivos (ambos para ES); anticentrómero en un sólo caso, con resultado negativo, y anti-RNP en 13 pacientes (21%), con todos los resultados negativos.
En cuanto a estudios de gabinete, se realizaron serie esofagogastroduodenal (SEGD), ultrasonografías, tomografías computarizadas, espirometrías y pletismografías. Del total de pacientes, se realizó SEGD a 56 (90%), de los que 12 presentaron RGE grado 1, 4 presentaron RGE grado 2 y 2 presentaron RGE grado 3. La espirometría se realizó en 33 pacientes (53%), 15 con resultado normal, 10 con restricción leve, 4 con patrón mixto, 3 con obstrucción leve y uno con restricción moderada. La pletismografía se realizó en 24 pacientes (39%), 8 con resultado normal, 6 con restricción leve, 5 con obstrucción leve, 4 con restricción moderada y uno con patrón mixto.
Se realizaron biopsias a 29 pacientes, 27 biopsias fueron de piel, una biopsia renal y un hemangioma rectal. Los hallazgos más característicos en la piel fueron la falta de anexos, la atrofia, el infiltrado inflamatorio por linfocitos y la colagenización.
La capilaroscopia se realizó solamente en un paciente con ES, observándose vasos dilatados.
TratamientoTodos los pacientes recibieron tratamiento inmunosupresor, utilizando un fármaco o una combinación de varios fármacos.
El medicamento más utilizado para la enfermedad fue metotrexato en 60 casos (96%) seguido de D-penicilamina en 9 casos (14%), esteroides en 7 casos (11,5%) y talidomida, cloroquina y ciclosporina A en un caso cada uno.
En su mayoría, el tratamiento de elección fue el de metotrexato sólo desde el inicio (43 casos). En los demás casos, se empezó con D-penicilamina, esteroides, talidomida, cloroquina o ciclosporina A que se asociaron a metotrexato durante la evolución hasta la sustitución por este último, a excepción de 2 pacientes que continuaron con D-penicilamina con buena evolución.
En cuanto al fenómeno de Raynaud asociado, la mayoría de los pacientes recibió nifedipina (14 casos) sin complicaciones y en 2 casos no se utilizó medicamento alguno para éste.
De los 62 pacientes con esclerodermia, a 13 (21%) se les realizó cirugía, en 12 pacientes (19%) el procedimiento quirúrgico consistió en lipoinyección con grasa autógena y solamente en un caso se realizó funduplicatura de Nissen debido a RGE grave.
El tiempo de tratamiento fue de 6 meses a 10 años de acuerdo con el tiempo de seguimiento en la clínica.
En cuanto a la evolución, se documenta en los expedientes clínicos mejoría en 40 pacientes (65%), 27 de estos corresponden a EL y 13 a ES. Los pacientes se dieron de alta por edad (18 años): 8 pacientes (12,5%) en buen estado general; 2 pacientes (3%) fueron trasladados a otros hospitales, 10 pacientes (16%) abandonaron el tratamiento y 5 pacientes (8%) se encuentran actualmente sin tratamiento. De los 62 casos, se constatan 3 pacientes (todos con ES) con importantes secuelas generales, con clase funcional 4. Asimismo, no se registraron casos de defunción.
DiscusiónEste estudio representa una muestra importante de niños con esclerodermia en México. Al igual que otras enfermedades autoinmunitarias, es más frecuente en las mujeres, lo que se relaciona con lo reportado en otros estudios6,8,9.
Afecta a los niños durante la etapa escolar y la adolescencia, con una media de edad de 8 años y con un rango que predomina entre 7 y 11 años, lo que coincide con estudios previos8,9.
El subtipo más comúnmente encontrado fue el de EL (71%) seguido por ES (29%), de acuerdo con estudios anteriores, aunque se describe una diferencia de hasta 1:10 entre la ES y la EL; no obstante, en este trabajo se encontró una diferencia de hasta 1,8:4,47.
Al momento del diagnóstico, la duración de la enfermedad fue a menudo prolongada, con una edad media de presentación entre la primera manifestación y el diagnóstico de 1,9 años y con un rango que variaba entre 0 y 10 años, sin mucha diferencia con lo reportado en la literatura médica, tanto en ES con una media de 2,4 años (rango de 0 a 10) como en EL con una media de 1,6 años (rango de 0 a 16,7). Este hallazgo, que ya se ha informado por otros autores, muestra que debe hacerse más esfuerzo para aumentar la consciencia de esta enfermedad entre los médicos y los profesionales de la salud en relación con el conocimiento de esta enfermedad.
Las lesiones en piel más frecuentes corresponden a morfea lineal seguida de morfea mixta y de morfea circunscripta, lo que coincide con otros reportes. El término morfea lineal de tipo golpe de sable se refiere a una lesión lineal generalmente ubicada en el cuero cabelludo frontoparietal o en la región frontal paramediana, que a menudo se asemeja al golpe de una espada y puede prolongarse hasta la cara. El síndrome de tipo Parry-Romberg o atrofia hemifacial idiopática se refiere a la atrofia hemifacial de la piel y del tejido debajo de la frente, por lo general sin participación cutánea importante10. Llama la atención que se encontraron 6 casos con ambas afecciones.
A diferencia de otros estudios5,8,11, en la ES se documentó involucro visceral y fenómeno de Raynaud con una mayor frecuencia, con afección respiratoria y gastrointestinal en todos los pacientes y fenómeno de Raynaud en el 88% de los casos.
Por lo general, en los pacientes con ES se desarrolló una atrofia progresiva de la piel que puede estar asociada a su adelgazamiento y, por consiguiente, presentaban menor puntuación en la escala del test de Rodnan. Sin embargo, esta medición tiene que validarse en los niños, que muestran una mayor puntuación (incluso en materia de salud), de acuerdo con el índice de masa corporal y la etapa de Tanner. No se puede discutir este punto, ya que no se utilizó esta medición en ninguno de los pacientes.
Por otra parte, Pavlov et al han demostrado que los cambios en la capilaroscopia reflejan las diferentes etapas de la enfermedad en pacientes con esclerosis sistémica12. Desafortunadamente, no se puede realizar medición al respecto, ya que esto se realizó en un solo paciente, debido a la falta de personal con experiencia en esto.
Dentro de los reportes encontrados en la literatura médica al igual que en los adultos en los niños con ES se ha comprobado la presencia de ANA con una frecuencia del 81 al 97% y en niños con EL con una frecuencia del 42,3%, menor que en los adultos5,7,8. Sin embargo, en este trabajo no se pudo realizar esta comparación, ya que se encontró este estudio solamente en el 56% de los casos, al igual que los anticuerpos anti-Scl70 y anticentrómero que se estudiaron sólo en 19 niños y en un paciente, respectivamente. Esto, debido a la falta de recursos económicos.
De acuerdo con las últimas recomendaciones, el tratamiento se enfoca a la afección predominante y, como en otros estudios, la afección dermatológica fue la más frecuente seguida del fenómeno de Raynaud, utilizándose en su mayoría metotrexato y nifedipina para ambas afecciones con buena respuesta aparente5,7–11.
El resultado de 5 años de supervivencia para este grupo fue del 100%, lo que coincide con informes anteriores de tasas de supervivencia7–9 y parece ser mejor que la supervivencia de los pacientes en los decenios anteriores. Sin embargo, la esclerodermia puede llegar a ser una enfermedad radical con evolución impredecible que requiere diagnóstico temprano y tratamiento oportuno, por lo que es importante continuar con el seguimiento de estos pacientes, ya que todos los pacientes con ES presentan diferentes grados de afección en diferentes órganos y, respecto a los niños con EL, no se conoce a ciencia cierta la posibilidad de progreso a la ES.
Se concluye que en España no varían las características clínicas de la esclerodermia en niños. Sin embargo, es importante recalcar la necesidad de un mayor conocimiento de esta enfermedad en medicina de primer contacto para el diagnóstico rápido y el tratamiento oportuno. Además, es importante superar la carencia de recursos en países en desarrollo para estudios auxiliares necesarios tanto para la clasificación como para el seguimiento.
Autor para correspondencia.
Zoilo Morel
Dirección: zoiloma@hotmail.com


