

El propósito de esta revisión es presentar los principales aspectos del componente genético de las enfermedades reumatológicas autoinmunes, incluyendo las características del modelo de herencia multifactorial o poligénico y sus formas monogénicas, así como los principales genes asociados en ambos casos. De igual manera, los cambios epigenéticos implicados, además de la influencia del ambiente y el sexo para conferir mayor riesgo a las mujeres de padecer alguna de estas enfermedades. Finalmente, se comenta acerca de los avances logrados por el estudio de las ciencias ómicas, abriendo camino a una nueva clasificación molecular de estas enfermedades, y así dirigirlo a una medicina personalizada. Se revisó la literatura de los últimos 5 años de publicaciones en lengua inglesa mediante la base de datos PubMed, se incluyeron 28 artículos de revisión y 19 artículos originales. Se discutió el papel de los factores genéticos que participan en la etiología de las enfermedades reumatológicas autoinmunes, gracias a la disponibilidad de estudios moleculares, lo que permite mayor comprensión de la fisiopatología y la posibilidad de realizar en un futuro cercano un diagnóstico y tratamiento basado en marcadores moleculares.
The purpose of this review is to present the main aspects of the genetic component of autoimmune rheumatic diseases, including the characteristics of the multifactorial or polygenic inheritance model, and its monogenic forms, as well as the main associated genes in both cases. The epigenetic changes involved, and the influence of the environment and sex that confer greater risk to women suffering from any of these diseases. Finally, to make known the advances that the study of omic sciences has allowed, opening the way to a new molecular classification of these diseases, aimed at personalized medicine. A review of the literature of the last 5 years, of English-language publications, in the PubMed database was performed and 28 review articles, and 19 original articles were included. Knowledge of the genetic factors involved in the aetiology of autoimmune rheumatic diseases, thanks to the availability of molecular studies, allows a better understanding of their pathophysiology and the possibility of diagnosis and treatment based on molecular markers in the future.
Las enfermedades reumatológicas autoinmunes, en su mayoría, se consideran poligénicas o con herencia multifactorial, dado que en su etiología están involucrados factores genéticos y ambientales; actualmente, los avances en el estudio del genoma humano han permitido conocer las variantes del genoma asociadas con estas enfermedades, con la visión de generar una nueva taxonomía, que permita reclasificarlas dependiendo de sus características moleculares y así predecir manifestaciones clínicas para prevenir y disminuir la morbimortalidad elevada en estas enfermedades1. El objetivo de esta revisión es presentar un panorama general sobre los componentes genéticos de las principales enfermedades reumatológicas autoinmunes.
MétodosSe revisaron publicaciones en revistas indizadas, en lengua inglesa a partir de la base de datos PubMed, abarcando un período de 2015 a 2019. Las palabras clave utilizadas para la búsqueda fueron enfermedades autoinmunes, genética, epigenética y sexo, la revisión se realizó en la biblioteca digital de la Benemérita Universidad Autónoma de Puebla. Para la redacción del presente trabajo se consideraron dentro de los fundamentos éticos las normas antiplagio.
Se revisaron 70 artículos, de los cuales se seleccionaron 47 por presentar los datos más relevantes en relación al tema, de estos, 28 fueron de revisión y 19 fueron originales.
DesarrolloHerencia multifactorial de las enfermedades autoinmunesLa mayoría de las enfermedades autoinmunes pertenecen a las enfermedades con herencia poligénica o multifactorial, ya que en su etiología intervienen alteraciones en múltiples genes, con algunas excepciones en las que se han descrito casos monogénicos; las alteraciones dadas en estos genes confieren susceptibilidad al individuo, el cual tras la exposición a determinados factores ambientales desarrolla la enfermedad2.
Respecto a la contribución de los factores genéticos en relación con los factores ambientales en una enfermedad multifactorial, en particular las enfermedades reumatológicas autoinmunes, se utiliza el término heredabilidad; así, por ejemplo, se ha determinado una heredabilidad en el lupus eritematoso sistémico (LES) del 43,9% y en la artritis reumatoide (AR) del 43,5%, lo que significa que en ambas enfermedades aproximadamente la mitad de la varianza fenotípica es explicada por factores genéticos3,4. Otro aspecto importante a considerar es la concordancia entre gemelos, en el LES se ha reportado una concordancia entre gemelos monocigotos del 24% y en gemelos dicigotos del 2%, lo cual confirma la importancia del componente genético, pues entre más genes se compartan mayor probabilidad de que 2 individuos emparentados presenten la misma enfermedad5. El estudio familiar de los pacientes con enfermedades reumatológicas autoinmunes permite observar que existe agregación y coagregación familiar, cuando en una misma familia existen varios casos con la misma enfermedad autoinmune o varios integrantes de la familia se encuentran afectados, pero la enfermedad autoinmune es diferente en cada uno de ellos. En los individuos que cuentan con un familiar de primer grado con AR se ha reportado un riesgo relativo (RR) de 5,28 y un intervalo de confianza del 95% (IC 95%): 4,60-6,07, un RR de 2,91 y un IC 95% de 2,49-3,42 para el LES y un RR de 3,13 y un IC 95% de 2,50-3,93 para el síndrome de Sjögren (SS) primario4. Por otro lado, un mismo individuo puede presentar más de una enfermedad autoinmune, lo cual se explica por el hecho de que las diferentes enfermedades de este grupo comparten hasta el 69% de sus loci de riesgo1.
El riesgo de presentar una enfermedad autoinmune también incrementa dependiendo del grado de parentesco y del sexo del familiar afectado. Se reporta que es mayor el riesgo entre familiares de primer grado y disminuye conforme se aleja el grado de parentesco, el riesgo también se verá incrementado si el caso índice es del sexo menos frecuentemente afectado; debido a que en la mayoría de las enfermedades reumatológicas autoinmunes el sexo más frecuentemente afectado es el femenino, si el caso índice corresponde a un varón, el riesgo será mayor; en el caso de la AR, el riesgo de padecer AR en un familiar de primer grado de un individuo afectado se incrementa 5 veces, en comparación con el riesgo de un individuo de la población general, y en caso de que el familiar afectado con AR sea del sexo femenino el RR es de 5,21 y el IC 95% de 4,47-6,08, mientras que si es varón el RR incrementa a 6,12 y el IC 95% a 4,90-7,654.
Genes de susceptibilidadGenes HLAEl complejo principal de histocompatibilidad major histocompatibility complex (MHC), localizado en el locus 6p21.3 está formado por un grupo de genes altamente polimórficos. Un polimorfismo de un solo nucleótido; single nucleotide polymorphism (SNP) es un cambio en la secuencia de nucleótidos que se encuentra en más del 1% de la población, por lo que se considera como una variante normal4. Lo que significa que los genes del MHC presentan múltiples alelos diferentes posibles lo que da identidad a cada individuo, el cual posee una combinación única para estos alelos; la versión humana de estos genes se conoce como human leucocyte antigens (HLA) y está formado por 3 regiones principales, la región HLA clase I está formado por los genes HLA-A, HLA-B y HLA-C; la región HLA clase II, contiene los genes HLA-DP, HLA-DQ y HLA-DR; y la región HLA clase III, contiene los genes del complemento C2, C4A, C4B (vía clásica) y FB (vía alterna), además de los genes para el factor de necrosis tumoral (TNF) y de la linfotoxina LTA y LTB entre otros, todos ellos importantes en la respuesta inflamatoria característica de la inmunidad innata6.
Los genes de la región HLA clase I se expresan en todas las células nucleadas y tienen como función reconocer y presentar antígenos que provienen del interior de las células a los linfocitos T CD8+ citotóxicos, mediante un receptor de célula T y su correceptor CD8, lo que desencadena la muerte de la célula infectada que expresa el antígeno. Por su parte, los genes de la región HLA clase II se expresan en la superficie de las células profesionales presentadoras de antígeno, para mostrar epítopes que provienen del medio extracelular a los linfocitos T CD4+ cooperadores, para lo cual requiere la interacción con un receptor de célula T y el correceptor CD4, de esta manera se desencadena la liberación de citocinas, la activación de macrófagos y de linfocitos B, estos últimos con la capacidad de producir anticuerpos7.
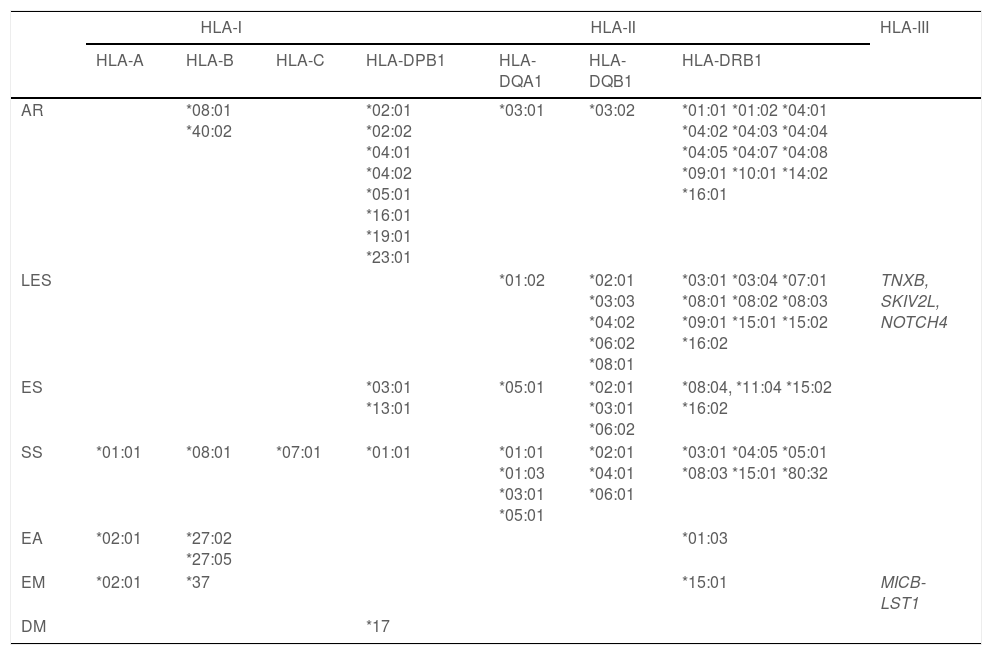
Las variantes en los genes del MHC representan el principal componente de la heredabilidad en las enfermedades autoinmunes. Existen alelos de riesgo, principalmente a nivel de los genes HLA clase II. La tabla 1 muestra los principales alelos de riesgo para varias enfermedades autoinmunes.
Variantes del MHC asociadas a enfermedades autoinmunes
| HLA-I | HLA-II | HLA-III | ||||||
|---|---|---|---|---|---|---|---|---|
| HLA-A | HLA-B | HLA-C | HLA-DPB1 | HLA-DQA1 | HLA-DQB1 | HLA-DRB1 | ||
| AR | *08:01 *40:02 | *02:01 *02:02 *04:01 *04:02 *05:01 *16:01 *19:01 *23:01 | *03:01 | *03:02 | *01:01 *01:02 *04:01 *04:02 *04:03 *04:04 *04:05 *04:07 *04:08 *09:01 *10:01 *14:02 *16:01 | |||
| LES | *01:02 | *02:01 *03:03 *04:02 *06:02 *08:01 | *03:01 *03:04 *07:01 *08:01 *08:02 *08:03 *09:01 *15:01 *15:02 *16:02 | TNXB, SKIV2L, NOTCH4 | ||||
| ES | *03:01 *13:01 | *05:01 | *02:01 *03:01 *06:02 | *08:04, *11:04 *15:02 *16:02 | ||||
| SS | *01:01 | *08:01 | *07:01 | *01:01 | *01:01 *01:03 *03:01 *05:01 | *02:01 *04:01 *06:01 | *03:01 *04:05 *05:01 *08:03 *15:01 *80:32 | |
| EA | *02:01 | *27:02 *27:05 | *01:03 | |||||
| EM | *02:01 | *37 | *15:01 | MICB-LST1 | ||||
| DM | *17 | |||||||
La disponibilidad de estudios moleculares como los estudios de asociación del genoma humano, genome wide association studies (GWAS), que permiten el análisis de hasta un millón de SNP en individuos afectados y no afectados de una enfermedad para comparar la frecuencia alélica en ambos grupos y de esta manera identificar alelos de riesgo, han permitido identificar tanto a genes HLA, como a otros genes no HLA asociados a diferentes enfermedades autoinmunes, la mayoría de estos genes están relacionados con la respuesta inflamatoria, la función de células B y T, la fagocitosis, la apoptosis y la producción de interferón gama8.
A la fecha se han realizado GWAS de las enfermedades reumatológicas autoinmunes más frecuentes, reportando aproximadamente 300 loci* de riesgo. La AR y el LES son los padecimientos con mayor número de loci asociados, probablemente porque en estas enfermedades se han realizado un mayor número de GWAS1. A continuación, se describen algunos genes que han mostrado variantes o alelos de riesgo para la presencia de diferentes enfermedades autoinmunes.
Interferon regulatory factor 5 (IRF5): localizado en la región 7q32.1, se ha asociado al LES y a la AR, codifica para un factor de transcripción llamado factor 5 regulatorio de interferón, cuya función es controlar la expresión de citocinas proinflamatorias; en los individuos con las variantes de riesgo, se incrementa la expresión del gen. Se encuentra fuertemente asociado al LES en poblaciones de Latino América9.
Interferon regulatory factor 7 (IRF7): se codifica en la región 11p15.5, está asociado al LES y a la esclerosis sistémica (ES), codifica un factor de transcripción del interferón tipo I (IFN-I)10.
Protein tyrosine phosphatase, non receptor-type 22 (PTPN22): se codifica en la región 1p13.2, está asociado al LES, a la AR y a la ES; codifica para una proteína tirosin fosfatasa intracelular linfoide específica, que controla la transducción de señales en los linfocitos T y B; los alelos de riesgo generan mayor concentración de interferón alfa (IFN-α) y niveles más bajos del TNF, además de que se han asociado con la producción de anticuerpos anti-DNA de doble cadena11.
Signal transducer and activator of transcription (STAT4): se codifica en la región 2q32.2-q32.3, está asociado al LES, a la AR, a la ES, a la esclerosis múltiple (EM) y al SS; codifica para una proteína transductora de señal y factor de transcripción que puede ser activado por las interleucinas 12 y 23; además estimula la producción de IFN- γ además de que es fundamental en la via de señalización del IFN-I; las variantes de riesgo incrementan la respuesta a la señal del IFN-α12.
B-cells caffold protein with ankyrin repeats 1 (BANK1): se codifica en la región 4q24, se asocia al LES, codifica para una proteína de andamio de células B con repeticiones de ankirina 1, reduce la producción del IFN-I por las células B; las variantes de riesgo alteran esta supresión, aumentando la función de este tipo celular13.
B lymphocytekinase (BLK): se codifica en la región 8p23.1, está asociado a la AR, al LES, a la ES y al SS, codifica una proteína tirosina cinasa, que se expresa en los linfocitos B, su función es la transducción de señales específicas de su linaje14.
Fc gamma receptor (FCGR): se codifica en la región 1q23.3 comprende una familia de receptores que se unen a la porción Fc de la inmunoglobulina G (IgG), se clasifican en receptores de alta afinidad (FCGR1A, FCGR1B y FCGR1C) y baja afinidad (FCGR2A, FCGR2B, FCGR2C, FCGR3A y FCGR3B); dentro de los de baja afinidad, FCGR2B es el único con función inhibitoria ya que el resto activan el receptor SNPs en el gen FCGR2B. Se han reportado como factor de riesgo para el LES y la AR, además de ser útiles para predecir la respuesta a terapia con anticuerpos monoclonales tales como rituximab15. La secuencia de los genes FCGR2C, FCGR3A y FCGR3B presentan variación en el número de copias, conocidos como copy number variations (CNV) lo cual confiere susceptibilidad tanto de las deleciones como de las duplicaciones, se ha reportado que deleciones en FCGR3B incrementa el riesgo de la AR y del LES16.
Neutrophil cytosolic factor (NCF): codifica para un componente del complejo NADPH oxidasa. Variantes en los genes NCF2 (1q25.3) y NCF1 (7q11.23) disminuyen la función de los mismos con la disminución de especies reactivas de oxígeno reactive oxygen species (ROS), que regulan la actividad de los macrófagos, para prevenir la inflamación, por lo que se han asociado con la AR, el LES y la EM9,17.
El incremento de los niveles del IFN-I y de los genes asociados con su expresión es un mecanismo fisiopatológico común a varias enfermedades reumatológicas autoinmunes que se ha denominado la señal de interferón y se observa principalmente en las enfermedades asociadas con la presencia de anticuerpos antinucleares, como el LES, el SS, la ES, la dermatomiositis (DM) y la enfermedad mixta del tejido conectivo (EMTC)18.
Factores epigenéticosLos genes descritos previamente han mostrado variantes a nivel de la secuencia de nucleótidos, aumentando o disminuyendo la función de los mismos, sin embargo, la expresión de un gen puede ser modificada por cambios que no afectan la secuencia de nucleótidos, sino que ocurren sobre alguna base o las histonas de la cromatina, así como por la acción de RNA no codificantes como los micro RNA (miRNA) y los RNA no codificantes largos, a estos cambios se les conoce como epigenéticos, los cuales pueden ser reversibles y heredables. Las modificaciones epigenéticas se han visto implicadas en el aumento de la respuesta inmune, generadas principalmente mediante la contribución de factores ambientales, contribuyendo a la fisiopatología de las enfermedades reumatológicas autoinmunes19.
La metilación del DNA consiste en la adición de un grupo metilo sobre la base nitrogenada citosina a través de las enzimas DNA metiltransferasas (DNMT), estas citosinas se localizan antes de una guanina en la misma cadena de DNA, existen regiones de más de 300 pares de bases que presentan alta densidad de estas secuencias por lo que se les denomina islas CpG, localizadas principalmente en la región promotora de los genes, sin embargo también pueden metilarse citosinas que no son parte de un dinucleótido CpG. Existen varias DNMT, como las DNMT2, DNMT3A, DNMT3B y DNMT3L que se encargan de la metilación de novo, mientras que la DNMT1 mantiene la metilación preexistente20.
Las histonas pueden sufrir diversos cambios, entre ellos acetilación, metilación, ubiquitinación, fosforilación y sumoilación; en el proceso de acetilación, mediante las acetiltransferasas de histonas, se transfieren un grupo acetilo al aminoácido lisina con lo que se activará la expresión del gen en cuestión, las desacetilasas de histonas al eliminar el grupo acetilo llevan a la inactivación del gen; por otro lado, las histonas pueden ser metiladas o desmetiladas, reacciones catalizadas por las metiltransferasas y desmetilasas de histonas, respectivamente; los aminoácidos de las histonas que aceptan estas marcas son la lisina y la arginina de las histonas H3 y H4, con diferentes efectos en la expresión genética, por ejemplo, cuando se agregan 3 grupos metilo a la lisina 4 de la histona 3 (H3K4me3) se incrementa la expresión del gen al aumentar su transcripción, por el contrario, si se agregan 2 grupos metilo a la lisina 9 de la histona 3 (H3K9me2) se inhibe la transcripción del gen21.
Los RNA no codificantes también tienen un papel importante en la regulación de la expresión génica, dependiendo del número de nucleótidos que los formen se clasifican en miRNA si tienen menos de 200 nucleótidos, generalmente de 21 a 25, o RNA no codificantes largos con 200 o más nucleótidos; en general los miRNA inhiben la transcripción de genes blanco o disminuyen la estabilidad del RNA mensajero (mRNA) evitando su traducción22.
Cambios epigenéticos en el lupus eritematoso sistémicoLos principales cambios epigenéticos reportados en pacientes con LES incluyen hipometilación en regiones promotoras de genes asociados con autoinmunidad, expresados en células T CD4+, lo que lleva a su sobreexpresión. La hipometilación es generada por inhibición de DNMT1, debido a varios mecanismos, uno de ellos es la acción de miRNAs; entre estos miR-148a y miR-126, ambos sobre expresados en células T CD4+ en pacientes con LES22.
Otros genes en los que se ha observado hipometilación están relacionados con la sobreproducción de interferón, entre ellos KIR en las células T. La sobreexpresión de IL-4 e IL-6 también se asocia a hipometilación de las islas CpG en pacientes con LES; varias enzimas reguladoras de la acetilación y desacetilación de histonas, están sobreexpresadas en células T CD4+ de pacientes con LES activo, entre ellas Sirtuin 1 (SIRT1), un tipo de deacetilasa de histona. La sobreexpresión de genes de citocinas como TNF-α e IL-17 en el LES se asocian a incremento de la acetilación de histonas; el nivel de expresión de miR-146a está disminuido en células mononucleares de sangre periférica de pacientes con el LES y correlaciona de manera inversa con la actividad de la enfermedad; la expresión de miR-15a en células B reguladoras muestra una correlación positiva con niveles séricos de autoanticuerpos anti-DNA de doble cadena (anti-dsDNA) en un modelo de ratón de lupus, mientras que miR-155 se correlaciona significativamente con la proteinuria y la actividad de la enfermedad en el LES23.
Cambios epigenéticos en la artritis reumatoideEn la AR se han documentado modificaciones epigenéticas a nivel de los fibroblastos sinoviales, que muestran hipometilación secundaria a una baja expresión de DNMT; por otro lado, el gen para CD40L está sobreexpresado en las células T CD4+, los fibroblastos sinoviales y el tejido sinovial presentan sobreexpresión de miR-155 y miR146, respectivamente, y altos niveles de IL-6, todo ello se asocia con sobreexpresión de miR-203 en los fibroblastos sinoviales22. A nivel de células mononucleares de sangre periférica se han reportado altos niveles de miR146 asociados con el incremento de citocinas proinflamatorias y TNF-α, mientras que la sobreexpresión de miR155 se asocia a valores elevados de proteína C reactiva, TNF-α, IL1β, tasa de eritrosedimentación y actividad de la enfermedad24.
Cambios epigenéticos en la esclerosis sistémicaEn la ES existen bajos niveles de metilación del DNA, a nivel de los promotores de los genes de CD40L, CD11a y CD70, lo cual resulta en sobreexpresión de los mismos; por otro lado, la hipermetilación en la región promotora del gen Forkhead box P3 (FOXP3), disminuye la proliferación de las células T reguladoras y conduce a niveles altos de JMJD3 en células T CD4+, lo anterior se asocia con la disminución de la metilación en H3K2725.
Cambios epigenéticos en el síndrome de SjögrenEn el SS se ha reportado disminución de la metilación en células epiteliales de glándulas salivales y en células T CD4+; además de la participación de múltiples miRNA, incluyendo a miR146 y miR155, que se han encontrado sobreexpresados en linfocitos T y células epiteliales de glándulas salivales de pacientes con el SS26,27.
Herencia monogénica en las enfermedades reumatológicas autoinmunesSe han descrito casos de enfermedades reumatológicas autoinmunes, como en el LES y en la AR, en donde la etiología no resulta de la asociación de variantes en múltiples genes, sino por el contrario, se deben a mutaciones en un solo gen. Una mutación, a diferencia de un polimorfismo, se presenta en menos del 1% de la población y es un cambio en la secuencia de nucleótidos que generalmente afecta el fenotipo o la función y causa una enfermedad; en su mayoría presentan herencia mendeliana, es decir, autosómico dominante, autosómico recesiva o ligada al cromosoma X28.
Los principales genes capaces de causar el LES monogénico son:
Deoxyribonuclease I-like 3 (DNASE1L3): se codifica en la región 3p14.3, mutaciones homocigotas en este gen son responsable del LES autosómico recesivo de inicio temprano, que cursa con anticuerpos anti-dsDNA y complemento bajo; el gen codifica para una endonucleasa, necesaria para la degradación de la cromatina durante la apoptosis, por lo que ante la deficiencia de esta enzima las micropartículas de cromatina son blancos de autoanticuerpos2,29.
Three prime repair exonuclease 1 (TREX1): se codifica en la región 3p21.31, mutaciones heterocigotas originan lupus sabañón familiar, con herencia autosómico dominante; codifica para una exonucleasa, que degrada fragmentos cortos de DNA, por lo que, ante una deficiencia de esta enzima secundaria a la mutación, estos fragmentos de DNA se acumulan y estimulan la producción de IFN I y con ello la autoinmunidad30.
SAM and HD domain containing protein 1 (SAMHD1): se codifica en la región 20q11.23, codifica una hidrolasa que degrada desoxinucleótidos trifosfatos a nivel intracelular, su deficiencia origina la producción de IFN I, como respuesta al acumulo de DNA no degradado, causando también lupus sabañón familiar, autosómico dominante31.
Otros genes asociados con el LES monogénico participan en la vía del complemento (C1Q, C2, C3 y C4), la apoptosis, la auto tolerancia (FAS, FASLG y PRKCD) y la degradación de ácidos nucleicos (RNASEH2A, RNASEH2B y RNASEH2C)32.
Influencia de los factores ambientales en el genomaLos factores ambientales que favorecen la expresión de las enfermedades reumatológicas autoinmunes en un individuo predispuesto genéticamente actúan a diferentes niveles, muchos de ellos directamente sobre el genoma, principalmente induciendo hipometilación. Por ejemplo, la radiación ultravioleta B (UVB), asociada al LES, induce disminución de la actividad de DNMTs causando hipometilación de las células mononucleares de sangre periférica, en especial las células T CD4+, con un aumento en la producción de IL1 y TNF-α. Niveles urinarios elevados de cadmio, elemento presente en el humo de cigarrillo y la dieta, se encontraron hasta 7 veces mas altos (7,4ug/l) en comparación con los no expuestos (1,0g/l) (p<0,001) y se asociaron con la hipometilación de genes que codifican para MGMT (0-6-metilguanosina DNA metiltransferasa). El cinc y el selenio son componentes de enzimas antioxidantes, en un estudio en pacientes con LES se encontraron niveles más bajos de los mismos (p=0,001) en comparación con controles sanos, el cinc en particular incrementa la metilación del DNA, por lo que su deficiencia contribuye a la hipometilación característica en este grupo de pacientes; de igual modo la silica favorece el desarrollo del LES, de la AR y de la esclerodermia al disminuir la metilación de DNA, y un ejemplo más es la infección por virus de Epstein-Barr también asociado a hipometilación de las islas CpG33,34.
Influencia del sexoLas enfermedades reumatológicas autoinmunes son más frecuentes en mujeres, lo cual se ha relacionado con factores hormonales y con la dosis de los genes presentes en los cromosomas sexuales. La proporción entre mujeres y varones en LES es de 9:135; en AR es de 4:1 y de 2:1 antes de los 50 y después de los 60 años, respectivamente36; para el síndrome antifosfolípido (SAF) se ha reportado de 5:1 en la presentación secundaria, asociada a otra enfermedad reumatológica autoinmune, y de 3,5:1 en la presentación primaria37; en SS primario se ha reportado de 14:1 (frecuencia [femeninos]=0,93; IC 95%: 0,89-0,96)38; y en la ES la relación es de 3-4:139.
Cromosoma XEl componente de cromosomas sexuales en la mujer es XX, mientras que en el varón es XY, lo cual representa la presencia de doble dosis de los genes del cromosoma X en el sexo femenino, para compensar este exceso de dosis, en la mujer, uno de los 2 cromosomas X se inactiva al azar en el desarrollo embrionario, formando una masa densa de heterocromatina, denominada corpúsculo de Barr, sin embargo, aproximadamente entre el 15 al 25% de los genes, escapan a la inactivación del X, lo que significa que se expresarán en doble dosis, mientras que los que se inactivan solo se expresarán en una dosis, la del alelo presente en el cromosoma X activo40.
Varios genes asociados a la respuesta inmune se encuentran en el cromosoma X, entre ellos Interleukin 9 receptor (IL9R), Interleukin 2 receptor gamma (IL2RG), los cuales codifican para los receptores de interleucina 9 y 2, respectivamente41. Toll-like receptor 7 (TLR7) escapa a la inactivación del X y junto con toll-like receptor 8 (TLR8) se asocia a un incremento en la producción de IFN-α en las mujeres con LES. C-X-C motif chemokine receptor 3 (CXCR3) y CD40 ligand (CD40LG) están hipometilados y sobreexpresados en las células T CD4+, mientras que Forkhead box p3 (FOXP3) está sobreexpresado en las células T CD8+ de pacientes con LES42.
Por otro lado, existen evidencias de la influencia del número de cromosomas X como factor de susceptibilidad a enfermedades reumatológicas autoinmunes que predominan en el sexo femenino, llama la atención la baja frecuencia de estas enfermedades en las pacientes con síndrome de Turner, quienes tienen solo un cromosoma X (45,X) y un incremento del riesgo para los varones con diagnóstico de síndrome de Klinefelter, que presentan uno o más cromosomas sexuales X extras (47,XXY; 48,XXXY; 49,XXXXY), siendo mayor el riesgo a mayor número de cromosomas X extras43.
Se ha reportado que la prevalencia para el LES en los varones con síndrome de Klinefelter es hasta de 14 veces más elevado en comparación con los varones con cariotipo 46,XY, incluso se han reportado casos de varones con LES con un cariotipo 46,XX. Cabe mencionar que si bien las pacientes con síndrome de Turner tiene menor riesgo por solo tener un cromosoma X, existen excepciones, como es el caso de las pacientes con mosaico 45,X/46,XX/47,XXX, ya que presentan células con 2 y hasta 3 cromosomas X, confiriendo mayor riesgo para LES y SS, o mujeres con cariotipo 47,XXXX, quienes presentan 2.5 veces más riesgo para el LES en comparación con las mujeres con cariotipo 46,XX44,45.
Las pacientes con síndrome de Turner por isocromosoma de brazos largos del cromosoma X(46,X,i(X)(q10)) presentan mayor riesgo de enfermedades autoinmunes ya que presentan un cromosoma X normal más otro cromosoma X que carece de brazos cortos y solo está formado por 2 brazos largos, lo que significa que hay triple dosis de los genes presentes en el brazo q del cromosoma X46.
Hormonas sexualesLas hormonas sexuales tanto endógenas (estrógenos, progesterona, andrógenos, prolactina) como exógenas (anticonceptivos orales y terapia de reemplazo hormonal) modifican la respuesta del sistema inmune; en general, se considera que los estrógenos tienen una función proinflamatoria y los andrógenos tienen función antiinflamatoria, lo que hace que la mujer sea más susceptible a las enfermedades reumatológicas autoinmunes. Las células del sistema inmune presentan receptores para estrógenos y andrógenos que a su vez activan factores de transcripción; en el caso de los estrógenos, inducen sobreexpresión de IRF5, incrementando los niveles de IFN1 y aumentan la expresión de TLRs intracelulares en las células mononucleares de sangre periférica (PBMCs); la testosterona por su parte, inhibe la proliferación de linfocitos y la actividad de las células Natural Killer; la progesterona inhibe la diferenciación de Th1 y Th17 y estimula la respuesta a Th2; y la prolactina incrementa la supervivencia de células B autoreactivas36,41.
Los niveles de las hormonas sexuales son variables entre las diferentes etapas de la vida, lo cual influye en la edad de inicio o en la gravedad de presentación de la enfermedad; en el caso del LES, afecta principalmente a las mujeres en edad reproductiva y el embarazo puede incrementar la actividad de la enfermedad, al aumentar la respuesta a Th2 y la producción de autoanticuerpos; el uso de anticonceptivos orales, la terapia de reemplazo hormonal y la menarca temprana se consideran factores de riesgo para el LES34. Por el contrario, en la AR se ha observado un pico de incidencia en la etapa posmenopáusica, principalmente en la quinta década de la vida mientras que la menopausia prematura, antes de los 45 años, se considera un factor de riesgo para la AR. Se ha observado una disminución de la actividad de la enfermedad con el embarazo, y aumento de la incidencia de la AR en los 3 primeros meses posparto (OR: 5,6; IC 95%: 1,8-17,6), asociado a un incremento de la prolactina y disminución de la progesterona; también se ha encontrado asociación entre el uso de anticonceptivos orales con la presencia de anticuerpos antiproteínas citrulinadas, anticitrullinated protein antibodies (ACPA), positivos (OR: 1,7; IC 95%: 1-1-2-6) y un incremento de la conversión de andrógenos a estrógenos por la aromatasa, activada por los mediadores de la inflamación36.
Perspectivas futurasEl avance en las técnicas moleculares y la bioinformática están haciendo posible la obtención, captura y análisis de una gran cantidad de datos de utilidad para la generación de biomarcadores que permiten la estratificación de los pacientes en grupos y el diseño de ensayos clínicos para brindar un tratamiento personalizado a los pacientes que padecen alguna de estas enfermedades reumatológicas autoinmunes; el proyecto PRECISESADS (Molecular Reclassification to Find Clinically Useful Biomarkers for Systemic Autoimmune Diseases) es un proyecto de la Unión Europea que inició en el año 2014 y tuvo como objetivo reclasificar las diferentes enfermedades sistémicas autoinmunes con base en sus características moleculares a partir de estudios de genómica principalmente mediante GWAS; transcriptómica, mediante la secuenciación de RNA en sangre periférica y tejidos específicos; epigenómica mediante análisis de metilación y proteómica mediante espectrometría de masas, cromatografía liquida, medición de citocinas y autoanticuerpos1.
Muestra de lo anterior, son los estudios realizados por Alarcón-Riquelme et al., quienes formaron grupos de pacientes con LES en base al porcentaje de neutrófilos y linfocitos los cuales se relacionaron con la actividad de la enfermedad mediante Systemic Lupus Erythematosus Disease Activity Index (SLEDAI); posteriormente en un análisis exploratorio haciendo uso de la transcriptómica y la plataforma CLUE, que permite el análisis de señales de expresión genética inducida por fármacos, evaluaron la capacidad de varios fármacos de revertir estas señales y observaron que los inhibidores de mTOR revirtieron la señal en el subgrupo dirigido por linfocitos, mientras que los inhibidores de TNF revirtieron la señal en el subgrupo de neutrófilos; se requerirán estudios experimentales de farmacodinamia para corroborar en cada grupo la efectividad de éstos fármacos. Los resultados podrían dirigir decisiones terapéuticas con objeto de brindar a cada paciente un tratamiento personalizado47.
ConclusionesEl conocimiento de los factores genéticos que participan en la etiología de las enfermedades reumatológicas autoinmunes permite una mayor comprensión de la fisiopatología de cada una de ellas. En el caso de las presentaciones poligénicas o multifactoriales, los genes HLA de clase II contribuyen principalmente con la heredabilidad de la enfermedad, sin embargo, polimorfismos en otros genes no HLA como IRF5, IRF7, PTPN22, STAT4, BANK1, BLK, FCGR y NCF también contribuyen de manera importante, principalmente mediante incremento en la expresión de interferón. En el caso del LES se han descrito formas monogénicas con herencia mendeliana autosómica dominante debida principalmente a mutaciones en los genes TREX1 y SAMHD1; y una forma autosómica recesiva por mutaciones en el gen DNASE1LB.
En relación con las alteraciones epigenéticas, el principal mecanismo implicado es la hipometilación del DNA, lo que confiere sobreexpresión de genes relacionados con la respuesta inmune. El componente de cromosomas sexuales se considera otro factor de riesgo para el sexo femenino, dado que entre el 15 al 25% de los genes escapan a la inactivación del X, lo que representa un exceso de dosis génica de varios loci asociados con autoinmunidad en comparación con el sexo masculino, entre ellos TLR7 que incrementa las concentraciones de IFN-α, así como una susceptibilidad dada por el componente hormonal, principalmente por estrógenos que en general tienen efecto proinflamatorio. El avance en las técnicas moleculares y la bioinformática permite entender mejor la fisiopatología y el manejo de una gran cantidad de datos lo que deriva en una clasificación molecular de estas enfermedades y con ello se otorgue un tratamiento específico a cada paciente.
FinanciaciónEsta investigación no ha recibido ninguna subvención específica de organismos de financiación, del sector público, comerciales o sin ánimo de lucro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


