



El presente documento refleja el posicionamiento del Colegio Mexicano de Reumatología y de expertos sobre el uso de medicamentos biocomparables (conocidos como biosimilares en otros países) en enfermedades reumáticas. En resumen, este posicionamiento considera que si bien los biocomparables deben considerarse como intercambiables, no es ética la sustitución automática de medicamentos sin previo aviso en pacientes estables durante el seguimiento; que la aprobación de un biocomparable debe llevarse a cabo solo después de revisar exhaustivamente las pruebas preclínicas y clínicas señaladas por la ley mexicana; que debe modificarse la forma de enfatizar en su nomenclatura que se trata de un medicamento biotecnológico innovador o biocomparable de manera clara; que no es adecuado elegir como tratamiento un biocomparable basándose únicamente en aspectos económicos ni realizarse la extrapolación de indicaciones basándose únicamente en la aprobación obtenida por el innovador y en ausencia de datos de seguridad y eficacia para el biocomparable.
The present document is a position statement of the Mexican College of Rheumatology on the use of biosimilars in rheumatic diseases. This position considers that biosimilars should be considered as interchangeable, that automatic substitution without previous notice in stable patients during follow-up is not ethical, that the approval of a biosimilar should only be given after exhaustive review of preclinical and clinical data marked by Mexican regulations, that it should be clearly stated in the nomenclature of biologic drugs which is the innovator and which is the biosimilar, that it is not correct to choose a biosimilar as treatment based only on economic reasons or extrapolate indications based only on the approval of the innovator and in the absence of safety and efficacy data for the biosimilar.
Con el advenimiento de los fármacos biotecnológicos se ha producido un parteaguas en la historia del tratamiento de padecimientos crónicos incapacitantes e incluso potencialmente mortales, como en el caso de la oncología, donde los fármacos biológicos, en casos particulares, ofrecen menos toxicidad y mayor supervivencia que los tratamientos convencionales; en lo que respecta a la reumatología, el éxito ha sido mucho más evidente en enfermedades tales como la artritis reumatoide (AR) o la espondiloartritis, donde un porcentaje considerable de pacientes alcanza la remisión clínica, condición que en el siglo pasado solo se lograba en menos del 20% de los casos1; adicional a esto se hace evidente la reintegración de los pacientes a sus actividades laborales, lo que representa menos gastos para las instituciones de salud. Actualmente, nos estamos enfrentando a la pérdida de la patente de algunos medicamentos innovadores y, con esto, a la llegada de los denominados medicamentos biosimilares, que en México son denominados biocomparables. De acuerdo con lo establecido en la normativa, un medicamento biocomparable es aquel fármaco biotecnológico no innovador que ha demostrado ser biocomparable en términos de seguridad, calidad y eficacia al medicamento biotecnológico de referencia a través de las pruebas establecidas en el marco regulatorio nacional. A nivel internacional, estos medicamentos son conocidos como medicamentos biosimilares2. Cabe mencionar que los medicamentos biocomparables son fármacos biológicos en sí mismos y la diferencia principal radica en que los innovadores crearon el producto y cuando se perdió la patente se realizaron réplicas de la formulación.
La introducción de los medicamentos biocomparables se acompaña de la posibilidad de reducir el costo de los tratamientos antirreumáticos e incrementar su disponibilidad en el momento en el que expiren las patentes de los medicamentos de referencia, con base en el supuesto de su menor costo3. Una de las ventajas actuales de estos productos es que, al haberse demostrado la biocomparabilidad de un medicamento, tienen la posibilidad de obtener aprobación sanitaria para las mismas indicaciones terapéuticas autorizadas para el medicamento innovador4, siempre y cuando cuente con la evidencia científica que permita la extrapolación de estas indicaciones. Los medicamentos biocomparables están disponibles en la Unión Europea desde el año 20064 (con la aprobación de Omnitrope®, somatropina), en Estados Unidos desde 2015 (con la aprobación de Zarzio®, filgrastim) y en México desde 2014 (con la aprobación de Zarzio®, filgrastim).
Actualmente, dentro de las opciones terapéuticas en enfermedades reumáticas, solo existe un biocomparable aprobado para su comercialización en México; dicho producto, identificado como CT-P13, es biocomparable de infliximab y le fue aplicada la legislación vigente en materia de biocomparabilidad. El medicamento biocomparable es conocido como Remsima®; no obstante, numerosos biocomparables están siendo desarrollados en la actualidad y pronto habrá más en el mercado. El marco regulatorio al cual se sujetan los biocomparables ha sufrido modificaciones, por ejemplo, la introducción de la NOM-257-SSA1-2014, en materia de medicamentos biotecnológicos, que en su última versión incluye ahora, como requisito para su aprobación, la presentación de pruebas de caracterización y estudios clínicos en pacientes, descritas también en otras normas actualizadas y guías de referencia5. Algunos medicamentos biotecnológicos en Reumatología se han encontrado con barreras de acceso para su aceptación y prescripción por parte de los médicos, propiciadas en parte por la introducción de medicamentos biotecnológicos autorizados antes del actual marco regulatorio nacional. El Colegio Mexicano de Reumatología (CMR) emite una actualización de su posicionamiento con respecto a los biocomparables que fue publicada por primera vez en 20126, a la luz de la nueva evidencia científica.
DefinicionesEn México, como se mencionó anteriormente, los medicamentos biocomparables se definen como los fármacos no innovadores que han demostrado características de calidad, eficacia y seguridad comparables al medicamento de referencia6,7. Existen otros medicamentos llamados biotecnológicos8,9 aún sin clasificar, cuyos registros sanitarios fueron otorgados antes del actual marco regulatorio de medicamentos biotecnológicos biocomparables; para estos casos la Norma Oficial Mexicana NOM-257-SSA1-20145 establece que tales medicamentos deberán cumplir con un proceso de revisión de sus registros sanitarios en apego al artículo 157 del Reglamento de Insumos para la Salud, a fin de cumplir con los requisitos que establece la Ley General de Salud10.
El término comparabilidad se refiere al proceso de la evaluación técnica de los atributos de calidad y el impacto de los cambios en el proceso de manufactura en las diferentes etapas del desarrollo de un biotecnológico o después de la aprobación del producto, por medio de pruebas analíticas y clínicas que demuestren que la calidad, la eficacia y la seguridad son equivalentes a las del medicamento de referencia11–14. Equivalencia se refiere al hecho de que el medicamento biocomparable cumple con la composición cualitativa y cuantitativa comparable al medicamento de referencia, proporciona el mismo efecto terapéutico15,16 y cumple con altos estándares de comparabilidad con el producto de referencia aprobado por las autoridades17.
La seguridad clínica de los medicamentos biotecnológicos no se evalúa solamente en términos de eventos adversos, también involucra la evaluación de su potencial inmunogénico, definido como la capacidad que una molécula tiene para provocar una respuesta inmune adaptativa celular o humoral de largo plazo y que genera memoria inmunológica18,19. Además de la farmacocinética, la farmacodinamia, la seguridad y la eficacia, que son evaluadas en la mayoría de los ensayos clínicos de biocomparables en el campo de la Reumatología, existen otras características de los medicamentos biocomparables que deben ser tomadas en cuenta para emitir una postura sobre su uso, y su definición se presenta a continuación. Trazabilidad se entiende como la capacidad para reconstruir la historia, recorrido o aplicación de un producto farmacéutico, identificando para ello el origen de sus componentes, los procesos aplicados al mismo y la distribución y la localización desde su producción y durante todo su «ciclo vital». Consiste en poder identificar las etapas que un producto va superando dentro de su proceso de fabricación (manipulaciones, composición, maquinaria empleada, temperatura a la que está sometido, número de lote, etc.) que se consideren de importancia y que pueden hacer variar el producto final para el consumidor20,21.
NomenclaturaActualmente, en México y otros países, los productos innovadores y los biocomparables se comercializan y distribuyen bajo la misma denominación común internacional de la sustancia activa. Por lo tanto, ambos productos comparten la misma clave asignada por el sector salud. Si los productos están dentro del cuadro básico, este proceso administrativo normado por la Secretaría de Salud bajo el Consejo de Salubridad General propicia que dichos productos puedan intercambiarse indiscriminadamente a razón de facilitar la oferta-demanda competitiva, o bien el abasto del medicamento. Sin embargo, lo anterior resulta problemático en términos clínicos, sobre todo en cuanto a la trazabilidad, que es un aspecto fundamental dentro del campo de la farmacovigilancia y que debería ser implementado por la totalidad de las personas físicas o jurídicas que intervengan en la cadena de producción, comercialización, distribución y dispensación del medicamento en cuestión (laboratorios, distribuidoras, operadores logísticos, farmacéuticos, médicos y enfermeras)20,21. Por ello, la Food and Drug Administration de los Estados Unidos publicó una propuesta para guiar la nomenclatura de productos biocomparables que no son propietarios del nombre original, y que en el momento de realizar esta revisión se encuentra en la etapa previa a su publicación oficial. En dicha propuesta se establece que actualmente es poco apropiado que los biocomparables compartan el nombre genérico o común internacional debido a que existe la necesidad de identificar claramente los productos biotecnológicos para hacer más eficiente la farmacovigilancia y diferenciar de forma precisa los productos que no hayan sido determinados como intercambiables22. En México, aún no contamos con propuestas oficiales para solucionar esta problemática actual, en beneficio del paciente. La Organización Mundial de la Salud ha propuesto añadir una serie de números y letras que sigan a la denominación común internacional mediante prefijos y sufijos clasificando las moléculas de una forma más precisa, pero esto no se ha adoptado universalmente. La European Medicines Agency (EMA) no ha dejado claro qué información debe ir incluida en la ficha técnica, aunque la posición europea es la de alinear biosimilares e innovadores en esta materia e identificarlos por una única denominación común internacional, por lo que añadir a esta un código o calificador iría en contra de esta idea, aunque es cierto que en otras partes del mundo, con mercados menos regulados, la idea ha tenido más aceptación. Esta idea ha sido promovida por la industria de los innovadores para entorpecer la entrada de biosimilares en el mercado.
Seguridad e inmunogenicidadLos datos clínicos de seguridad, incluyendo evaluaciones de inmunogenicidad, son necesarios para evaluar la biocomparabilidad23 por la probabilidad de pequeñas diferencias entre proteínas y la preocupación de que pudieran resultar en un incremento en la inmunogenicidad y la hipersensibilidad. El producto biocomparable puede también diferir del producto de referencia en la formulación, las impurezas, los excipientes y los componentes clínicamente inactivos, lo cual puede resultar en diferencias inmunogénicas clínicamente relevantes, lo suficientemente importantes como para negar la licencia a un producto que busca la denominación de biocomparable23. Tanto la Food and Drug Administration como la Organización Mundial de la Salud requieren de al menos un estudio de inmunogenicidad para comparar la respuesta inmune entre el biosimilar y el producto de referencia24–26. Adicionalmente, la EMA plantea que deberían realizarse estudios de seguimiento de inmunogenicidad y seguridad posteriores a su autorización27.
Intercambiabilidad y sustitución automáticaEn México, la normativa no establece que un biocomparable pueda ser considerado como intercambiable (Reglamento de Insumos para la Salud). No existe un posicionamiento por parte de la agencia regulatoria nacional (Comisión Federal para la Protección contra Riesgos Sanitarios [COFEPRIS]) sobre la intercambiabilidad o sustitución de los medicamentos biotecnológicos y su correspondiente biocomparable; sin embargo, la práctica de la sustitución automática a nivel institucional es indiscriminada, en parte debido a un claro vacío legal y a la falta de homologación y acuerdos entre las diferentes autoridades y actores clave en el mercado farmacéutico. Con la denominación de biocomparabilidad no debe asumirse la intercambiabilidad. El término intercambiabilidad se refiere a la designación que permite a un biocomparable ser prescrito en lugar del producto de referencia28. Ser sustituido sin la intervención del profesional de la salud que prescribió el producto de referencia se denomina sustitución automática2. Un producto biocomparable puede ser considerado como intercambiable con el producto de referencia solo cuando: 1) el producto cumple con la definición de biocomparable, y 2) se ha demostrado que tiene el mismo resultado clínico en el paciente después de cambiar o alternar la formulación, siendo esto valorado en un tiempo razonable29. Intercambiar la formulación (o hacer switch) se refiere a comenzar el tratamiento con el producto de referencia y posteriormente continuar el tratamiento con el biocomparable, o viceversa. La sustitución se refiere a iniciar el tratamiento con el producto de referencia, cambiar por el biocomparable y posteriormente regresar al producto de referencia, o comenzar con el biocomparable, continuar con el de referencia y terminar con el biocomparable29.
Adicionalmente, para asumir intercambiabilidad, el riesgo en términos de seguridad o disminución de la eficacia al cambiar o alternar entre el producto de referencia y el biocomparable no es mayor que el riesgo que tiene el paciente si solo se usa el producto de referencia sin hacer ningún cambio de producto o alternar entre el producto de referencia y el biocomparable30.
Sin embargo, la sustitución automática sí conlleva riesgo de falla en términos de farmacovigilancia, ya que no hay indicación médica de por medio, siendo una práctica llevada a cabo por el farmacéutico sin una adecuada trazabilidad2.
Extrapolación de indicacionesExtrapolación significa que una vez que se ha establecido la biocomparabilidad en una o más indicaciones, un biocomparable puede ser aprobado para algunas o todas las indicaciones adicionales para las cuales el producto de referencia o innovador tiene aprobación, sin necesidad de ensayos clínicos comparativos en las demás indicaciones31,32. En Estados Unidos, es posible autorizar un biocomparable para múltiples condiciones para las cuales el producto de referencia esté autorizado con base en evidencia para su uso en una indicación, siempre y cuando esté apoyado por una justificación científica23. Actualmente, en México no se cuenta con un documento oficial que señale las características para la extrapolación de indicaciones en biocomparables y se realiza a discreción de la autoridad, respaldada en parte por la opinión de comités de expertos. Por otro lado, la posibilidad de obtener la extrapolación de indicaciones es uno de los principales atractivos del desarrollo de biocomparables33; no obstante, es controvertido, dado que la fisiopatología de las enfermedades y los mecanismos de acción de los fármacos biotecnológicos no son necesariamente iguales en todas las dolencias ni con todos los tipos de medicamentos biotecnológicos. Cabe mencionar que no todos los países permiten la extrapolación de indicaciones; un ejemplo es la EMA27, que permite que los resultados de los ensayos clínicos llevados a cabo en pacientes con enfermedades reumáticas sean extrapolados a enfermedades inflamatorias del intestino, mientras que la Agencia de Salud de Canadá34 no lo permite33. Las consideraciones regulatorias para la extrapolación de indicaciones se presentan en la tabla 1.
Consideraciones regulatorias para la extrapolación de indicaciones en los biocomparables
| Health Canada34 | Un biosimilar puede solicitar autorización para todas las indicaciones aprobadas para el medicamento biológico de referencia autorizado en Canadá al que se hace referencia |
| La autorización de las indicaciones solicitadas depende de la demostración de similitud entre el biosimilar y el medicamento de referencia con base en datos derivados de estudios comparativos estructurales, funcionales, estudios no clínicos y clínicos. Cuando se ha establecido similitud, pueden darse indicaciones incluso si no se realizan estudios clínicos en cada indicación. Se debe proporcionar una justificación detallada que justifique científicamente la autorización del biosimilar en cada indicación teniendo en cuenta el mecanismo o mecanismos de acción, el mecanismo fisiopatológico de la enfermedad o las afecciones involucradas, el perfil de seguridad, el régimen de dosificación, la experiencia clínica con medicamento de referencia, y cualquier consideración caso por caso. Bajo ciertas condiciones se pueden requerir datos clínicos adicionales para soportar una indicación en particular | |
| EMA27 | Cuando la biosimilaridad se ha demostrado en una indicación terapéutica, la extrapolación de datos clínicos a otras indicaciones del producto de referencia podría ser aceptable, pero debe estar científicamente justificada. En los casos donde no esté claro si la seguridad y la eficacia confirmadas en una indicación serían relevantes para otra indicación, se requerirán datos adicionales. La extrapolación debe considerarse teniendo en cuenta la totalidad de los datos, es decir, la calidad, los datos no clínicos y clínicos. Se espera que la seguridad y la eficacia se puedan extrapolar cuando la comparabilidad biosimilar haya sido demostrada a través de estudios físico-químicos y estructurales exhaustivos, así como con pruebas funcionales in vitro complementadas con datos clínicos (eficacia y seguridad y/o datos PK/PD) en una indicación terapéutica |
| Se solicitarán datos adicionales en situaciones, como: | |
| 1. Casos donde el biofármaco (la sustancia activa del producto de referencia) interactúa con varios receptores que pueden tener un impacto diferente en las diversas indicaciones terapéuticas probadas y no probadas | |
| 2. Situaciones donde el biofármaco (sustancia activa) tiene más de un sitio activo y los sitios pueden tener un impacto diferente en distintas indicaciones terapéuticas | |
| 3. En los casos donde la indicación terapéutica estudiada no es relevante para los demás en términos de eficacia o seguridad, es decir, no es sensible para detectar diferencias en todos los aspectos relevantes de eficacia y seguridad. | |
| La inmunogenicidad está relacionada con múltiples factores, incluyendo la vía de administración, el régimen de dosificación, los factores relacionados con el paciente y los factores relacionados con la enfermedad (por ejemplo, tratamientos concomitantes, tipo de enfermedad, estado inmune). Por lo tanto, la inmunogenicidad podría diferir entre las diversas indicaciones terapéuticas. Por lo anterior, debe justificarse la extrapolación de la inmunogenicidad de la indicación/vía de administración estudiada hacia otros usos | |
| FDA24 | Si el medicamento cumple con los requisitos legales para obtener la licencia como un producto biosimilar bajo la sección 351 (k) de la Ley PHS con base, entre otras cosas, en datos derivados de un estudio o estudios clínicos suficientes para demostrar la seguridad, pureza y potencia en una condición apropiada de uso, se podrá solicitar la licencia/autorización para una o más condiciones adicionales de uso (indicaciones terapéuticas) para las cuales el producto de referencia tiene licencia. Se deberá proporcionar una justificación científica suficiente para extrapolar los datos clínicos que sustente la biosimilaridad para cada condición de uso solicitada |
| Dicha justificación científica para la extrapolación debe incluir, por ejemplo, las siguientes cuestiones para las condiciones de uso comprobadas y extrapoladas: | |
| • El/los mecanismo(s) de acción (MOA) en cada condición de uso para la cual se solicita autorización; esto puede incluir: | |
| - El antígeno/receptor(es) para cada actividad/función relevante del producto | |
| - La unión, la respuesta de dosis/concentración y el patrón de señalización molecular al asociarse/unirse el/los receptor(es) y los antígeno(s) | |
| - Las relaciones entre la estructura del producto y las interacciones entre el blanco/receptor | |
| - La localización y expresión del blanco/receptor(es) | |
| • La PK y la biodistribución del producto en diferentes poblaciones de pacientes (las medidas de farmacodinamia pertinentes también pueden proporcionar información importante sobre el MOA) | |
| • La inmunogenicidad del producto en diferentes poblaciones de pacientes | |
| • Las diferencias en las toxicidades esperadas en cada condición de uso y en la población de pacientes (incluyendo si las toxicidades esperadas están relacionadas con la actividad farmacológica del producto o con las actividades fuera del blanco) | |
| • Cualquier otro factor que pueda afectar la seguridad o la eficacia del producto en cada condición de uso y la población de pacientes para la cual se solicita la autorización | |
| Las diferencias entre las condiciones de uso con respecto a los factores descritos anteriormente no excluyen necesariamente la extrapolación. Una justificación científica debe abordar estas diferencias en el contexto de la totalidad de las pruebas que apoyan una demostración de biosimilaridad | |
| Al elegir la condición de uso que permitirá la posterior extrapolación de datos clínicos a otras condiciones de uso, la FDA recomienda que el patrocinador considere elegir una condición de uso que sea adecuadamente sensible para detectar diferencias clínicamente significativas entre los 2 productos | |
| Solo se puede obtener la autorización para una condición de uso que haya sido previamente autorizada para el producto de referencia |
El coste del tratamiento va más allá del precio con el que se oferta. Sabemos que un medicamento es competitivo no solo en eficacia y seguridad, sino también cuando tiene un precio justo y un valor agregado en relación con el beneficio que le otorga al paciente y a la institución de salud. La principal ventaja que debería tener el uso de biocomparables es la competitividad. En nuestro país, en enero de 2017 el biocomparable autorizado Remsima® tenía un coste de 7,370$/100mg, mientras que el medicamento innovador, Remicade®, tenía un coste de 10,405$/100mg; es decir, sí hay un beneficio económico en cuanto a coste del fármaco; no obstante, el biocomparable aún no cuenta con ningún programa de apoyo a pacientes, mientras que el innovador sí ofrece un extenso programa de apoyo a pacientes35.
Marco regulatorioLos requerimientos para obtener la aprobación como biosimilares por la Food and Drug Administration, Health Canada y la EMA incluyen estudios extensos tanto in vitro, demostrando la similitud con el medicamento de referencia en términos de atributos de calidad, como estudios preclínicos y clínicos demostrando la comparabilidad farmacocinética y de eficacia, seguridad e inmunogenicidad24,27,33. Actualmente en México, los medicamentos biocomparables deben sustentar su registro bajo las pruebas establecidas en las normas oficiales mexicanas 177 (2013), 257 (2014), 059 (2015), 164 (2015) y 220 (2015). También las leyes locales, los lineamientos y las guías de referencia mundial están en consonancia con 4 principios fundamentales propuestos por la Organización Mundial de la Salud en materia de biosimilares, que en resumen establece que: 1) deben presentarse estudios de caracterización fisicoquímica que demuestren que las heterogeneidades encontradas en la estructura química del fármaco biotecnológico biosimilar no impacten de manera negativa en la eficacia y la seguridad del tratamiento; 2) deben presentarse estudios de actividad biológica donde se demuestre, en modelos experimentales de relevancia, que el biosimilar tiene el mismo mecanismo de acción que el innovador; 3) deben presentarse estudios clínicos en pacientes que padecen la enfermedad en cuestión con una muestra poblacional representativa a fin de demostrar que el biosimilar es un equivalente terapéutico o no inferior al medicamento innovador, y 4) deben presentase estudios de farmacovigilancia que demuestren que el biosimilar no representa un riesgo de seguridad mayor que el medicamento innovador.
Revisión sistemática de la literaturaCon el objetivo de emitir la postura del CMR con base en la mejor evidencia disponible se realizó una revisión sistemática de la literatura concentrada en los ensayos clínicos de biosimilares cuyo uso se ha propuesto en enfermedades reumáticas. Se utilizaron los términos «biosimilar», «biosimilars», «infliximab», «adalimumab» y «etanercept» en la búsqueda realizada en las bases de datos de PubMed y Cochrane Library, utilizando la función de filtro para la búsqueda de ensayos clínicos aleatorizados. Se realizó una búsqueda manual para localizar estudios de los intentos de copia que estuvieron o están disponibles en México, como Kikuzubam® e Infinitam® (Kikuzubam® ya no está disponible en México, Infinitam®, sí). La figura 1 muestra el diagrama de flujo de las publicaciones revisadas en la revisión sistemática. La primera etapa de la revisión consistió en la revisión por título y resumen de los artículos encontrados; la revisión se hizo por duplicado por parte de 2 reumatólogos que eligieron los artículos que cumplían con los criterios de selección. Los desacuerdos fueron resueltos por una tercera persona. Los artículos que fueron seleccionados por título y resumen fueron posteriormente revisados en extenso por ambos revisores para determinar cuáles cumplían con los criterios de selección. Una vez identificados, se revisaron las referencias de estos para identificar nuevos títulos que pudieran ser de interés. Se revisó el texto completo de todos los artículos identificados por esta vía y se incluyeron los que cumplieron con los criterios de selección. Los criterios de selección fueron los siguientes: 1) publicaciones en inglés o español; 2) ensayos clínicos o estudios de extensión o registros nacionales; 3) evaluación de eficacia, seguridad, extensión de indicaciones, intercambiabilidad o inmunogenicidad; 4) uso en cualquiera de las siguientes enfermedades: AR, artritis idiopática juvenil, espondiloartritis, espondilitis anquilosante, artritis psoriásica, lupus eritematoso sistémico; 5) que incluyeran niños, adolescentes o adultos, y 6) publicados hasta junio de 2016. Los datos fueron extraídos y los resultados de la revisión fueron presentados a reumatólogos miembros del CMR en el segundo Simposio Internacional de Artritis Reumatoide, llevado a cabo del 29 de septiembre al 1 de octubre de 2016 en Puerto Vallarta (México). Durante esta reunión se presentaron los enunciados de la propuesta de postura ante los miembros, se sometió a votación y se discutieron los comentarios que surgieron durante el proceso de votación. Los miembros del CMR que no asistieron a la reunión fueron invitados a participar en la votación de la postura vía electrónica, en la cual también tuvieron oportunidad de hacer comentarios a la misma. En total, en la votación presencial y electrónica participaron 74 reumatólogos.
Resultados
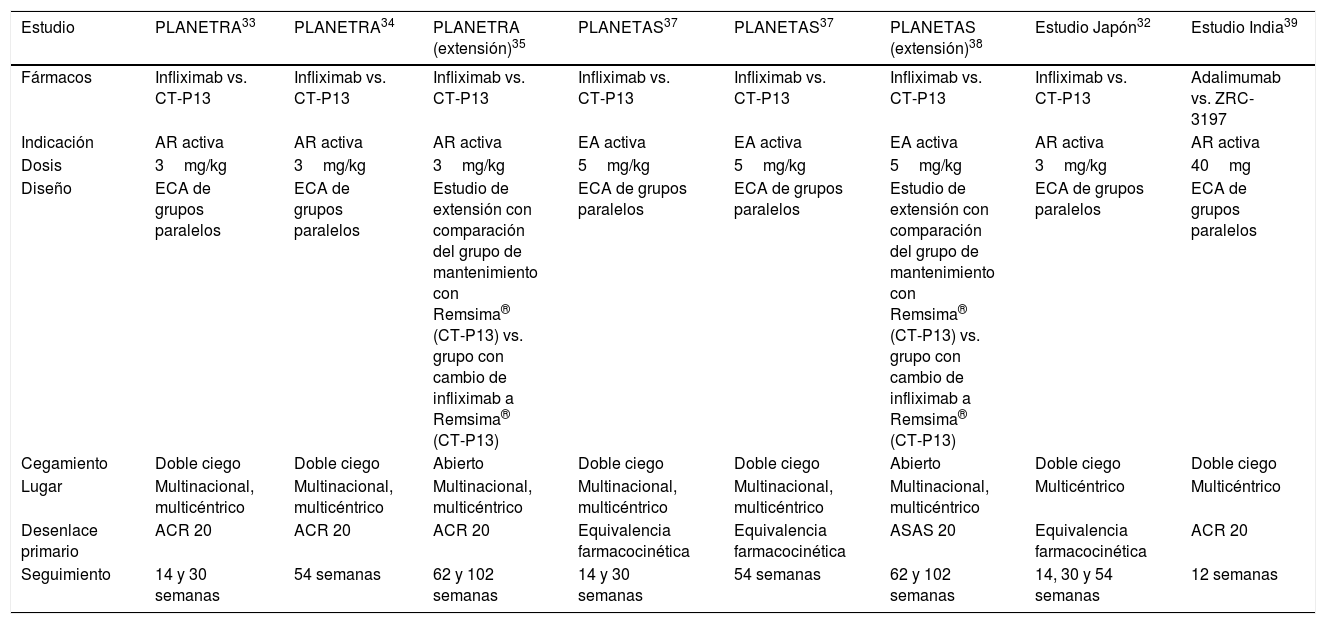
En la búsqueda sobre información de los productos biotecnológicos aún sin clasificar Kikuzubam® e Infinitam® no se obtuvo ningún resultado; no hay evidencia disponible en la literatura sobre la eficacia y seguridad de ninguno de estos productos. En la búsqueda sobre biosimilares en PubMed y Cochrane Library se obtuvieron 212 artículos; una vez eliminados los 85 títulos repetidos, permanecieron 127 artículos para revisión por título y resumen. Por esta vía se identificaron 109 que no fueron elegibles. Se evaluaron por texto completo 18; de estos, solo 5 cumplieron con los criterios de inclusión. Al revisar las referencias y los artículos relacionados se identificaron 3 trabajos más. En total se incluyeron 8 artículos. Las características generales de los estudios se encuentran en la tabla 2. De las 8 publicaciones incluidas, 7 fueron sobre el biocomparable de infliximab y una sobre el biocomparable de adalimumab; de las publicaciones relacionadas con el biocomparable de infliximab, una es un estudio realizado en Japón36, 3 publicaciones pertenecen al estudio multinacional PLANETRA37–39 y 3 al estudio multinacional PLANETAS40,41; la publicación del biocomparable de adalimumab fue un estudio multicéntrico realizado en India42. Todas las publicaciones comparan la molécula CT-P13 biosimilar de infliximab frente a la molécula original de infliximab y todas las investigaciones fueron financiadas por Celltrion, la empresa que desarrolló el biosimilar; no se obtuvieron resultados de biocomparables de etanercept. Los estudios PLANETRA y PLANETAS publicaron sus resultados a la semana 3037,40; luego, publicaron los resultados tras 54 semanas de seguimiento38,43, realizando posteriormente un estudio de extensión39,41 donde el grupo de pacientes con fármaco biocomparable permaneció con la misma terapia y el grupo que había sido tratado con el producto de referencia cambió al biocomparable a partir de la semana 62 y hasta la semana 102.
Características generales de los estudios incluidos en la revisión sistemática
| Estudio | PLANETRA33 | PLANETRA34 | PLANETRA (extensión)35 | PLANETAS37 | PLANETAS37 | PLANETAS (extensión)38 | Estudio Japón32 | Estudio India39 |
|---|---|---|---|---|---|---|---|---|
| Fármacos | Infliximab vs. CT-P13 | Infliximab vs. CT-P13 | Infliximab vs. CT-P13 | Infliximab vs. CT-P13 | Infliximab vs. CT-P13 | Infliximab vs. CT-P13 | Infliximab vs. CT-P13 | Adalimumab vs. ZRC-3197 |
| Indicación | AR activa | AR activa | AR activa | EA activa | EA activa | EA activa | AR activa | AR activa |
| Dosis | 3mg/kg | 3mg/kg | 3mg/kg | 5mg/kg | 5mg/kg | 5mg/kg | 3mg/kg | 40mg |
| Diseño | ECA de grupos paralelos | ECA de grupos paralelos | Estudio de extensión con comparación del grupo de mantenimiento con Remsima® (CT-P13) vs. grupo con cambio de infliximab a Remsima® (CT-P13) | ECA de grupos paralelos | ECA de grupos paralelos | Estudio de extensión con comparación del grupo de mantenimiento con Remsima® (CT-P13) vs. grupo con cambio de infliximab a Remsima® (CT-P13) | ECA de grupos paralelos | ECA de grupos paralelos |
| Cegamiento | Doble ciego | Doble ciego | Abierto | Doble ciego | Doble ciego | Abierto | Doble ciego | Doble ciego |
| Lugar | Multinacional, multicéntrico | Multinacional, multicéntrico | Multinacional, multicéntrico | Multinacional, multicéntrico | Multinacional, multicéntrico | Multinacional, multicéntrico | Multicéntrico | Multicéntrico |
| Desenlace primario | ACR 20 | ACR 20 | ACR 20 | Equivalencia farmacocinética | Equivalencia farmacocinética | ASAS 20 | Equivalencia farmacocinética | ACR 20 |
| Seguimiento | 14 y 30 semanas | 54 semanas | 62 y 102 semanas | 14 y 30 semanas | 54 semanas | 62 y 102 semanas | 14, 30 y 54 semanas | 12 semanas |
La farmacocinética y la farmacodinamia del biosimilar CT-P13 mostraron ser comparables con las del infliximab en todos los estudios. La eficacia fue medida por múltiples indicadores en las 7 publicaciones. El estudio PLANETRA37,38 evaluó la respuesta ACR 20, ACR 50, ACR 70, el índice simplificado de la actividad de la enfermedad (SDAI), la respuesta EULAR buena o moderada, la actividad baja o en remisión (definida por DAS28-CRP), la necesidad de terapia de rescate y la remisión ACR/EULAR, no encontrando diferencias significativas en las semanas 14, 30 y 54. El estudio de extensión39 tampoco encontró diferencias en ninguno de los indicadores en las semanas 62 y 102 entre los grupos de mantenimiento de CT-P13 y el grupo de cambio de infliximab a CT-P13. El estudio del biocomparable de adalimumab42 no evaluó ni la farmacocinética ni la farmacodinamia.
El estudio PLANETAS40 evaluó, a las semanas 14 y 30, ASAS 20, ASAS 40, cambio en ASDAS-CRP, cambio en BASDAI, cambio en BASFI y cambio en BASMI; en la semana 5443 de seguimiento evaluaron ASAS 20, ASAS 40, remisión parcial ASAS, cambio en ASDAS-CRP, cambio en BASDAI, cambio en BASFI y cambio en BASMI. No se observaron diferencias significativas entre los grupos con CT-P13 e infliximab. El estudio de extensión41 tampoco encontró diferencias en ninguno de los indicadores a las semanas 62 y 102 entre los grupos de mantenimiento de CT-P13 y el grupo de cambio de infliximab a CT-P13.
El artículo de Takeuchi et al.36 evaluó ACR 20, ACR 50, ACR 70, el porcentaje de respondedores EULAR, cambio en DAS28 (VSG), cambio en DAS28 (PCR), cambio en SDAI y en CDAI a las semanas 14, 30 y 54; solamente se observaron diferencias en los cambios en SDAI, siendo mayores en el grupo de CT-P13 que en el grupo de infliximab (−18,4±15,8 vs. −14,1±12,2, p<0,05). El artículo de Jani et al. evaluó ACR 20, ACR 50 y ACR 70 y tampoco encontró diferencias entre el innovador de adalimumab y su biocomparable ZRC-319742.
La seguridad fue evaluada por todos los estudios en sus fases de grupos paralelos. El porcentaje de eventos desfavorables y reacciones adversas presentados en al menos el 1% de los pacientes en el grupo de CT-P13 fue, respectivamente, de 64,8 y 34,3% en el estudio PLANETRA, 60,1 y 35,2% en el estudio PLANETAS y 88,2 y 84,3% en el estudio de Takeuchi et al. En el grupo de infliximab dichos porcentajes fueron de 63,9 y 26,3% en el estudio PLANETRA, 60,8 y 35,9% en el estudio PLANETAS y 86,8 y 88,1% en el estudio de Takeuchi et al. Ninguna de las diferencias fue estadísticamente significativa. En el estudio de Jani et al., durante el seguimiento de 12 semanas, de 60 participantes incluidos en cada grupo, se presentaron 13 eventos adversos en 7 sujetos del grupo tratado con el biocomparable de adalimumab, mientras que en el grupo del producto de referencia se presentaron 15 eventos adversos en 10 sujetos; estas diferencias no fueron estadísticamente significativas.
Los resultados de seguridad en los estudios de extensión son los siguientes: el estudio PLANETAS reportó que la proporción de pacientes que experimentaron por lo menos un evento adverso fue del 40,9% en el grupo de mantenimiento a diferencia del grupo que cambió del innovador a CT-P13, cuya proporción fue del 71,4%; el estudio de extensión PLANETRA reporta una proporción similar de eventos adversos (53,5 vs. 53,8%).
El estudio PLANETRA, llevado a cabo en pacientes con AR, reportó inmunogenicidad en la semana 14 para el grupo de CT-P13 e infliximab de 25,4 y 25,8%, para la semana 30, de 48,4 y 48,2%, y para la semana 54, de 49,1 y 48,3%, respectivamente. En el estudio de extensión el grupo de permanencia con CT-P13 y el grupo de cambio al biocomparable mostraron inmunogenicidad en la semana 78 en el 44,7 y 46,2% de los participantes, respectivamente, y en la semana 102, en el 40,3 y 44,8%; ninguna de las diferencias fue estadísticamente significativa. El artículo de Takeuchi et al.36, también realizado en pacientes con AR, reportó inmunogenicidad en la semana 14 en un 19,6% de los pacientes que recibían CT-P13 y un 15,1% de los que eran tratados con infliximab; en la semana 30, un 25,5 y un 26,4%, y a la semana 54, un 25,5 y un 32,1% presentaban inmunogenicidad, respectivamente. Ninguna de las diferencias fue estadísticamente significativa.
El estudio PLANETAS, hecho en pacientes con espondilitis anquilosante, reportó una inmunogenicidad a la semana 14 en el grupo de CT-P13 e infliximab del 9,1 y 11%, a la semana 30, del 27,4 y 22,5%, y del 19,5 y 23% en la semana 54, respectivamente. En el estudio de extensión se reportó inmunogenicidad en los grupos de permanencia y cambio a la semana 78 en el 23,3 y 29,8% de los participantes, y a la semana 102 en el 23,3 y 27,4%, respectivamente. Ninguna de las diferencias fue estadísticamente significativa. El estudio de Jani et al.42 reportó inmunogenicidad a la semana 12 de seguimiento en 2 de los 60 participantes del grupo del biocomparable de adalimumab y en un participante de los 60 incluidos en el grupo tratado con el producto de referencia.
DiscusiónDada la falta de evidencia científica sobre eficacia, seguridad, inmunogenicidad e intercambiabilidad de los productos biotecnológicos aún sin clasificar, como Kikuzubam® e Infinitam®, no es posible desarrollar una discusión alrededor de su uso. Los biosimilares pueden ofrecer una alternativa más económica que los productos biológicos existentes que han perdido ya sus patentes y favorecen una competencia limpia, mejorando, por tanto, el acceso de un mayor número de pacientes a los medicamentos biológicos, contribuyendo así a la sostenibilidad financiera de los sistemas sanitarios21; no obstante, la literatura del efecto de los medicamentos biocomparables en enfermedades reumáticas sigue siendo limitada. Si bien existen más estudios sobre farmacocinética, farmacodinamia, eficacia, seguridad e inmunogenicidad de los medicamentos biocomparables, muchos de estos han sido realizados en sujetos sanos44-47 o con otro tipo de padecimientos, como enfermedad inflamatoria intestinal o enfermedades cutáneas.
Con base en los resultados de los estudios incluidos en la revisión sistemática, podemos afirmar que existe suficiente evidencia para aceptar la comparabilidad farmacocinética y farmacodinámica entre CT-P13 y su producto de referencia, infliximab. Está reportado en otras fuentes que CT-P13 es idéntico en la secuencia de aminoácidos, la forma farmacéutica, la fuerza, la composición y la vía de administración al producto innovador48. Solo encontramos un estudio del biocomparable de adalimumab que evaluó eficacia, seguridad e inmunogenicidad en pacientes con AR, si bien la necesidad de estudios adicionales de seguridad y efectividad dependerá de factores como la incertidumbre que se tiene sobre el efecto del medicamento o el grado de entendimiento que se tiene sobre los mecanismos de acción del producto y de la fisiopatología de la enfermedad; para alcanzar la meta de demostrar que no existen diferencias clínicamente significativas entre el producto de referencia y el biocomparable, generalmente son necesarios por lo menos estudios de farmacocinética humana y evaluaciones de inmunogenicidad23.
Los resultados de eficacia de CT-P13 en AR y espondilitis anquilosante son comparables a los obtenidos con su producto de referencia; no obstante, el objetivo principal en los estudios de Takeuchi et al. y PLANETAS no fue la eficacia, sino la equivalencia farmacocinética; solamente el estudio PLANETRA tuvo como objetivo primario la evaluación de la eficacia por medio de la respuesta ACR 20, que a pesar de ser el indicador que solicitan las agencias internacionales para obtener la aprobación para su comercialización, no es el indicador de mayor relevancia clínica. Por lo tanto, debe considerarse que al no haber estudios diseñados para evaluar los indicadores clínicamente significativos de eficacia con CT-P13, no se puede asegurar que se tenga el poder estadístico suficiente para observar diferencias entre los grupos. El estudio de Jani et al.42 sí tuvo como objetivo principal la eficacia clínica medida con ACR 20, por lo que sí existe información que confirme que en términos de eficacia no existen diferencias entre el innovador y el biocomparable; no obstante, ACR 20 no es necesariamente el indicador de éxito del tratamiento que más se utiliza en la práctica clínica.
En ninguno de los estudios de grupos paralelos se observaron diferencias entre el grupo con el producto de referencia y aquel con el biocomparable; solamente en la extensión del estudio PLANETAS se observó una diferencia importante, ya que el grupo que cambió del innovador al biocomparable tuvo un incremento del 30% en la aparición de eventos adversos comparado con el grupo de mantenimiento con el innovador. El tamaño de la muestra de los estudios incluidos en esta revisión no permite determinar con claridad el perfil de seguridad de los esquemas de tratamiento. Se requieren estudios de farmacovigilancia para tener herramientas con las que se pueda llevar a cabo dicha evaluación.
Los resultados de inmunogenicidad, tanto en los estudios de grupos paralelos como en sus estudios de extensión con cambio del producto innovador al biocomparable, no muestran diferencias significativas entre los grupos; no obstante, llama la atención el alto porcentaje de participantes que desarrollan anticuerpos contra estos biológicos. Si bien la «humanización» de los anticuerpos monoclonales murinos ha mejorado ampliamente su tolerabilidad in vivo, algunos anticuerpos humanizados derivados de la secuencia, y hasta los que son «completamente» humanos, todavía acarrean potencial inmunogénico49. El alto porcentaje de inmunogenicidad no puede ser atribuido solamente al producto de referencia o al biocomparable, y se ha propuesto que la inmunogenicidad siempre va a estar presente cuando se trata del uso terapéutico de anticuerpos debido a la naturaleza de los sitios específicos de antígeno49. En el estudio PLANETRA, el 25% de los participantes habían desarrollado inmunogenicidad a la semana 14 y casi la mitad a la semana 30, en contraste en el estudio de Takeuchi et al.; a pesar de haber incluido también participantes con AR, obtuvo porcentajes de inmunogenicidad más bajos, siendo el más alto de ellos de 32,1%. Aun cuando una de las principales diferencias entre el estudio de Takeuchi et al. y el estudio PLANETRA es el grupo étnico incluido, esto no parece explicar el contraste en los resultados de los estudios, ya que una investigación comparó la sensibilidad étnica hacia los anticuerpos monoclonales en personas japonesas y no japonesas y encontró que la mayoría de los anticuerpos monoclonales siguen el comportamiento esperado independientemente de la etnicidad; la única excepción parece ser adalimumab, que sí mostró diferencias en su comportamiento entre grupos étnicos50. La respuesta inmunogénica podría entonces estar determinada más bien por la fisiopatología de la enfermedad. El estudio PLANETAS, que incluyó participantes con espondilitis anquilosante, reportó inmunogenicidad en un porcentaje bastante menor, no rebasando el 30%. Otros estudios han reportado una menor inmunogenicidad en pacientes con espondilitis anquilosante en comparación con pacientes con AR51. Lo anterior nos lleva a cuestionar la pertinencia de la extrapolación de indicaciones, dadas las diferencias observadas entre pacientes con AR y con espondilitis anquilosante y la falta de evidencia de su eficacia y seguridad en otras indicaciones. Hay que tener cierta cautela al manejar estos datos y utilizarlos de un modo comparativo y complementario, ya que se debe tener en cuenta la variabilidad de los resultados en función de la técnica diagnóstica utilizada. Es decir, si un cambio potencial en la inmunogenicidad después de 12 meses de tratamiento fuera el mayor incidente acontecido, para averiguar su relevancia clínica se requeriría de un laboratorio centralizado así como la correlación de los resultados con los niveles del medicamento en el organismo, respuesta al tratamiento e incidencia y severidad de las reacciones adversas21.
Los datos proporcionados en las etapas de extensión a 102 semanas de los estudios PLANETAS y PLANETRA muestran que cambiar el producto de referencia infliximab por su biocomparable CT-P13 no tiene efectos deletéreos en la eficacia, la seguridad o la inmunogenicidad comparado con el uso continuo de CT-P1352. Adicionalmente, una revisión de la literatura, que además de tomar en cuenta los estudios antes mencionados, incluyó datos de la vida real con pacientes con enfermedad inflamatoria intestinal, concluye que cambiar del producto de referencia a su biocomparable CT-P13 no tiene efectos indeseables52. No obstante, aunque contar con evidencia sobre el efecto del cambio del producto de referencia al biocomparable ciertamente es de mucha utilidad, esos datos no resuelven la pregunta sobre el efecto de la intercambiabilidad en la eficacia, la seguridad y la inmunogenicidad, ya que ninguno de los estudios hizo el cambio de vuelta al producto de referencia, condición que es requerida para evaluar la intercambiabilidad23. Cabe recordar que para demostrar que la sustitución del tratamiento es factible debe demostrarse previamente la intercambiabilidad. Al no encontrarse en la literatura estudios sobre el biocomparable de adalimumab que evalúen el efecto del cambio de terapia o intercambiabilidad, no puede establecerse si es seguro alternar el producto innovador y su biocomparable.
PosicionamientoEl CMR propone el siguiente posicionamiento ante los productos biocomparables en México:
- 1.
Que un biotecnológico innovador es aquel que ha sido desarrollado y registrado en el mundo por primera vez para una o más indicaciones y que un biocomparable es aquel que, utilizando las mismas técnicas de biología molecular, pretende lograr una estructura y función equiparables a las del producto innovador.
- 2.
Que la obtención de los productos biotecnológicos requiere de un proceso complejo que no garantiza que los fármacos biocomparables que no cumplan con los requerimientos regulatorios tanto nacionales como internacionales sean iguales a los innovadores, y debido a ello y a la falta de evidencia científica acerca de su eficacia, seguridad e intercambiabilidad, este tipo de biocomparables no deben considerarse como intercambiables.
- 3.
Que se debe tomar en cuenta lo siguiente: 1) en Reumatología los ensayos clínicos con biocomparables se han llevado a cabo en pacientes que nunca antes habían recibido terapia biológica; 2) que no hay evidencia de la seguridad y eficacia de su intercambiabilidad en pacientes estables con el producto de referencia, y 3) que la información de seguridad de los pacientes durante el seguimiento a largo plazo no puede evaluarse de manera adecuada si se cambia un medicamento innovador por un biocomparable o viceversa. No es correcto aceptar la práctica de sustitución automática de medicamento.
- 4.
Que cada producto debe enfatizar en su nomenclatura de manera clara que se trata de un medicamento biotecnológico innovador o biocomparable y debe darse a conocer su lote con el fin de asegurar la farmacovigilancia de todo medicamento y garantizar la seguridad de cada medicamento administrado al paciente.
- 5.
Que los involucrados con la producción, la prescripción y el uso de los medicamentos biotecnológicos están obligados al ejercicio de farmacovigilancia a largo plazo y que los eventos secundarios deben reportarse al portal de COFEPRIS (www.cofepris.gob.mx).
- 6.
Que no debería cambiarse a un paciente estable con el producto de referencia a un biocomparable solamente por razones económicas.
- 7.
Que, en los pacientes vírgenes a tratamiento biológico, la decisión sobre la terapéutica con los productos biológicos aprobados (de referencia o biocomparable) debe estar dictada por una evaluación individual del riesgo/beneficio y no basarse únicamente en aspectos económicos.
- 8.
Que no debe realizarse o aprobarse la extrapolación de indicaciones basándose únicamente en la aprobación obtenida por el producto de referencia ante COFEPRIS y en ausencia de datos de seguridad y eficacia para el biocomparable.
- 9.
Se recomienda a COFEPRIS, el Consejo de Salubridad General, las instituciones de salud, de economía y actores clave que establezcamos juntos un diálogo para determinar oficialmente la unificación de criterios de precios, intercambiabilidad y sustitución del tratamiento con el medicamento biocomparable, con la mejor evidencia científica disponible y en beneficio del paciente, encontrando así una vía para cumplir con el derecho a la protección a la salud especificada en nuestra constitución.
- 10.
Esta postura deberá actualizarse cada 2 años o según surja evidencia importante al respecto.
El CMR manifiesta que este grupo de trabajo está a favor del desarrollo de medicamentos biotecnológicos biocomparables, tanto en México como en otras partes del mundo, así como de su aprobación, sin interferencia externa, por las agencias reguladoras, siempre y cuando, además de demostrar su equivalencia farmacocinética, sean sometidos a los más altos estándares de calidad en términos de producción y desarrollo, que se sometan a una comparación de su eficacia y seguridad frente al producto de referencia por medio de estudios fase iii y iv con un adecuado poder estadístico, y que sean seguidos por un estricto programa de farmacovigilancia. Las metas para el desarrollo de biocomparables deben incluir: 1) un ahorro sustancial para las instituciones públicas de salud; 2) que los pacientes puedan adquirir estos medicamentos con pago de bolsillo; 3) favorecer el acceso de sectores más amplios de la población a estos medicamentos, y 4) no anteponer el factor económico a la eficacia terapéutica y la óptima seguridad de los pacientes.
NotaLas opiniones expresadas en este artículo pertenecen a los autores y solo refleja el posicionamiento del CMR y no la postura oficial o posicionamiento de ninguna de las instituciones a las que están adscritos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesNinguno de los autores recibió pago de ninguna especie por su participación en el presente artículo. Daniel Xibille declara ser asesor y ponente de Hospira-Pfizer, Eli Lilly, Pfizer, Glaxo Smith Kline, Janssen, Bristol y Abbvie. Sandra Carrillo y Leonardo Limón declaran ser asesores y ponentes de Eli Lilly, Pfizer, Roche, Janssen, Bristol, Abbvie, Glaxo Smith Kline y Novartis. Abdieel Esquivel declara haber sido patrocinado para consultorías, asesorías de marca, eventos académicos y conferencias nacionales e internacionales organizados por Pfizer, Novartis, Eli Lilly, Abbvie, Sanofi, Hospira, Roche, Janssen, Stendhal, Merck, UCB Pharma y MSD. Gabriela Huerta-Sil, Ramiro Hernández, Guadalupe Olvera-Soto, Luis Javier Jara-Quezada y Marcela Pérez-Rodríguez declaran no tener ningún conflicto de intereses.
Agradecemos a los doctores que participaron de forma presencial en la sesión de la postura: Dr. Francisco Javier Aceves Ávila, Dr. Jorge Enrique Aguilar Arreola, Dra. Guadalupe Cecilia Aguilar Domínguez, Dra. Lilia Andrade Ortega, Dra. Ana Cecilia Arana Guajardo, Dra. María de Jesús Araujo Arias, Dr. Marcos Daniel Ayala Arzola, Dr. Jorge Alberto Barragán Garfías, Dr. Mario Alfredo Chávez López, Dra. Lucia Comellas Kirkerup, Dr. José Arturo Covarrubias Cobos, Dra. Beatriz Alicia de Cortina Camou, Dr. Juan Carlos de la Cruz Castillejos, Dra. Karla Irene de la Cruz Rodríguez, Dr. Adrián Alejandro de la Madrid Cernas, Dr. José Noel Díaz Muñoz, Dra. Sandra Enciso Pelaéz, Dra. María Amparo Enríquez Maldonado, Dra. Grissel Espericueta Arriola, Dra. Diana Laura Ferrusquia Toriz, Dra. Sonia de la Mercedes Gacitúa Zambrano, Dr. Víctor Adán Gallaga Gutiérrez, Dr. Oscar Gallegos Hernández, Dr. José Luis García Figueroa, Dr. Conrado García García, Dr. César Leonardo García López, Dra. Imelda García Olivera, Dra. Karla Maritza García Osuna, Dr. Rafael García Rascón, Dra. Claudia Elizabeth Gómez López, Dr. Osvaldo González La Riviere, Dra. Mariana Paola González Mora, Dra. Leticia Gutiérrez Pérez, Dr. Jorge Carlos Guzmán Murillo, Dr. Jaime Hadid Smeke, Dr. Éufrates Hernández Núñez, Dra. María del Carmen Hernández Quiroz, Dr. Guillermo Fernando Huerta Yáñez, Dra. Graciela Ibáñez Landín, Dr. Jorge Jaimes Hernández, Dra. Xochitl Jiménez Jiménez, Dr. Bernardo Julián Martínez, Dra. Montserrat Lamuño Encorrada, Dra. Marysol Lendechy Velázquez, Dra. Luz Aurora León Legaría, Dra. Ana Laura Marines Castillo, Dra. Gloria Esther Martínez Bonilla, Dra. Lucia Verónica Maya Piña, Dra. Claudia Irene Melendez Mercado, Dra. Josefina Xóchitl Mendoza Vázquez, Dr. Juan Manuel Miranda Limón, Dr. Mauricio Montero Luna, Dr. Carlos Moya McClaugherty, Dra. Gracida Karol Mugica de la Lanza, Dr. Roberto Negrete López, Dr. Victoria Obiala Ezenwa, Dr. Francisco Olan, Dra. Liliana Ortiz Martínez, Dra. Leslie Osuna Garibaldi, Dra. Ana Virginia Perla Navarro, Dra. Rebeca Ramírez González, Dra. Greta Cristina Reyes Cordero, Dra. Jacqueline Reyes Rueda, Dr. Humberto Alfredo Ricardez Puente, Dra. Tatiana Sofía Rodríguez Reyna, Dra. Roxana Minerva Rodríguez Romo, Dr. Mauricio Eduardo Rubio Sánchez, Dr. Raymundo Ruíz García, Dr. Miguel Ángel Saavedra Salinas, Dr. Arturo Sánchez Arriaga, Dr. Antonio Sánchez González, Dra. Tania Sánchez Hernández, Dra. Maria del Lucero Sanchez Segura, Dra. Karina Santana de Anda, Dra. Clara Shumski Flaschner, Dra. Sandra Araceli Sicsik Ayala, Dr. Luis Humberto Silveira Torre, Dra. Claudia Magdalena Solís Alvarado, Dr. Leobardo Terán Estrada, Dra. Norma Edith Torres Gudiño, Dr. Ignacio Alfredo Valerio Morales, Dr. Lorenzo Valle Gómez, Dr. Julio Villagómez Calderón, Dr. Arturo Villarreal Ortega, Dr. Erick Adrian Zamora Tehozol y al resto de los colegiados que participaron en la votación vía electrónica.


