



Systemic juvenile idiopathic arthritis (sJIA) is a chronic childhood inflammatory disease. SJIA accounts for approximately 5–15 per cent of all cases of JIA and has a high morbidity and mortality rate. In this disease, pulmonary complications (PC) other than pleuritis are much less frequent and not easily recognised by clinicians. Pulmonary hypertension, the most severe PC, is associated with uncontrolled disease and use of biologic therapies. We present a case of a school-age female with sJIA who died of acute cardiopulmonary instability secondary to pulmonary venous-occlusive disease demonstrated by necropsy. We describe her clinical evolution. We also undertook a narrative review of the literature about PC in sJIA to discuss the current state of the art regarding this complication. High disease activity and the use of multiple therapies include disease-modifying anti-rheumatic drugs should be a red flag for clinicians when discounting PC and pulmonary hypertension. The combination of chest X-ray, electrocardiogram and echocardiogram appear to be the best tests to achieve an early diagnosis.
La artritis idiopática juvenil sistémica (AIJs) es una enfermedad juvenil crónica que representa aproximadamente del 5 al 15% de todos los casos de AIJ y tiene una elevada tasa de morbimortalidad. En esta enfermedad, las complicaciones pulmonares (CP) distintas a pleuritis son mucho menos frecuentes, y no fácilmente reconocibles por los clínicos. La hipertensión pulmonar, la CP más grave, está asociada a la enfermedad incontrolada y el uso de terapias biológicas. Presentamos el caso de una mujer en edad escolar con AIJs que falleció debido a inestabilidad cardiopulmonar aguda secundaria a enfermedad venosooclusiva confirmada en la necropsia. Describimos su evolución clínica, y también realizamos una revisión narrativa de la literatura relativa a CP en AIJs, para debatir los avances más recientes sobre esta complicación. La elevada actividad de la enfermedad y el uso de terapias múltiples que incluyen fármacos antirreumáticos modificadores de la enfermedad deberían servir de signo de alarma a los clínicos para descartar CP e hipertensión pulmonar. La combinación de placas de tórax, electrocardiograma y ecocardiograma parece ser el mejor conjunto de pruebas para lograr un diagnóstico precoz.
Systemic juvenile idiopathic arthritis (sJIA) is a chronic childhood inflammatory disease classified as a subtype of JIA, according to the International League of Associations of Rheumatology (ILAR) criteria. It is characterized by the presence of arthritis associated with systemic manifestations like fever, macular, salmon-pink rash, lymphadenopathy, hepatomegaly and/or splenomegaly and serositis. Also increase of acute phase reactants, hyperferritinemia and anemia are usually seen. The sJIA accounts for approximately 5–15 per cent of all cases of JIA with a high morbid-mortality. Recent findings have shown an excellent response to biologic therapies, particularly to interleukin 1 and 6 inhibitors.1,2
Macrophage activation syndrome (MAS) is the best known and described sJIA complication. Persistent fever, thrombocytopenia, aspartate transaminase (AST) elevation, hypertriglyceridemia and/or hypofibrinogenemia, appeared associated with hyperferritinemia in a patient with suspected or confirmed sJIA.3 On a different side, pulmonary complications (PC) are much less frequent and difficult to be recognized by clinicians. Pulmonary hypertension, the most severe PC, is associated with uncontrolled disease and biologic therapies requirements. It is necessary a high degree of suspicion for this complication. Delayed diagnosis compromises the beginning of specific pulmonary therapy and reduces the quality of life of these children.
We present a case of a school-age female suffering from sJIA who died of acute cardiopulmonary instability secondary to pulmonary veno-occlusive disease. We describe her clinical evolution since diagnosis until her death. Later, we realize a literature narrative review to discuss the current state of the art about this complication.
Case reportSix years old female with sJIA diagnosis at two years old. The disease first sign was a MAS episode. She improved with high dose of corticosteroids, methotrexate and anakinra. Two months later she suffered a new episode of MAS during corticosteroid tapering. After the resolution of this anakinra was changed to tocilizumab.
During the following months, she showed good evolution, allowing to decrease corticosteroids and withholding one year after the beginning of the disease. Later she presented again arthritis with intermittent low-grade fever. Corticosteroids were restarted and tocilizumab was replaced by canakinumab. After remission corticosteroids were suspended until recurrence eight months later. Corticosteroids controlled these symptoms always with doses over 0.5mg/kg/day.
During the previous weeks before her visit to the emergency department (ED) she referred asthenia and dyspnoea with the exercise. They were attributed to disease activity. An echocardiogram was informed as normal. A Chest ray performed during a respiratory infection did not show cardiomegaly.
She came to the ED after 24h of limbs pain, hypothermia and acrocyanosis. She felt discomfort with abdominal pain. After observation she was discharged with an appointment the next day with her paediatric rheumatologist. Six hours later, she returned to the ED. She had cyanosis, poor perfusion, shortness of breath and drowsiness. Also low blood pressure, tachycardia and severe metabolic acidosis. The patient received fluid therapy, broad-spectrum antibiotics and stress dose of corticosteroids. Despite this, she suffered a cardiorespiratory arrest. She was intubated and reanimated for 30min. After recovering she was transferred to pediatric intensive care unit (PICU).
The child arrived at PICU with pallor, poor perfusion, acrocyanosis and more than 10s of capillary filling. She had bilateral lung crackles, tachycardia without cardiac murmur and hepatomegaly. Pulmonary edema was observed thorax radiography (Fig. 1). Despite intensive care the patient suffered two more cardiorespiratory arrests. The first one was resolved with severe cardiac dysfunction seen by echocardiography. The second one did not respond to advance cardiopulmonary resuscitation. After 40min the medical team decided to stop. The parents accepted the necropsy. Days later it was informed as lung's pulmonary veno-occlusive disease (Fig. 2). The cultures and molecular tests were negative for bacterial, viral or fungal infections. The pulmonary complication was considered as the main cause of progressive hemodynamic instability and death.

 Pulmonary parenchyma showing two veins with marked intimal hyperplasia and patched thickening of alveolar septum (eosin hematosilin, 4×). (B) Detail of alveolar septshes demonstrating a thickening produced by a proliferation of capillary lights (eosin–hematosilin, 10×). (C) Immunohistochemical staining for CD31 that shows the proliferation of hair lights (CD31, 20×).")
(A) Pulmonary parenchyma showing two veins with marked intimal hyperplasia and patched thickening of alveolar septum (eosin hematosilin, 4×). (B) Detail of alveolar septshes demonstrating a thickening produced by a proliferation of capillary lights (eosin–hematosilin, 10×). (C) Immunohistochemical staining for CD31 that shows the proliferation of hair lights (CD31, 20×).
We describe the case of a patient with sJIA and pulmonary veno-occlusive disease that resulted in pulmonary artery hypertension and heart failure. Although diagnosis could not be made until a necropsy, we think that pulmonary hypertension was the main responsible for the haemodynamic instability, the sudden clinical deterioration and the subsequent death.
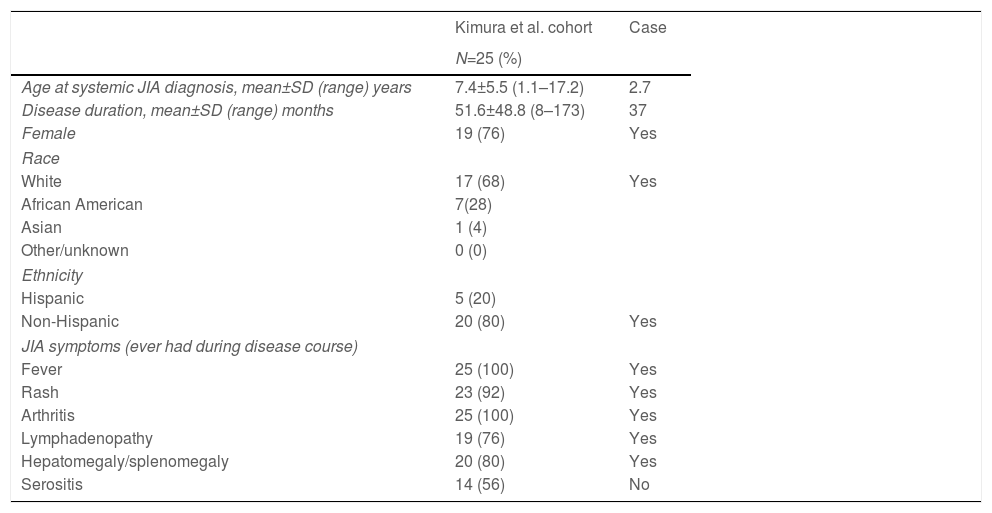
The PC in sJIA disease is uncommon but more frequent than initially thought. Kimura et al. described in 2013 an international cohort of 25 sJIA patients who developed pulmonary hypertension, alveolar proteinosis or interstitial lung disease and compared their clinical and demographic data with 389 sJIA patients that were part of the Childhood Arthritis and Rheumatology Research Alliance (CARRA) registry.4 As in our case, sJIA patients with PC were more likely to be female, had more systemic manifestations and had been exposed to multiple therapies (Tables 1 and 2). Almost all children were exposed to more than one drug, and nearly three out of four had received one or more disease-modifying anti-rheumatic drugs (DMARD). The most common biologic DMARD taken when pulmonary symptoms developed was an IL-1 inhibitor (anakinra, Table 2). A possible causal relationship between exposure to these medications and the development of PC was suggested, taking into account that most cases were diagnosed after the year 2004, coinciding with the most common use of these drugs for sJIA treatment.
Patient demographics and systemic JIA disease and pulmonary disease characteristics in the Kimura et al. cohort and in the patient case.
| Kimura et al. cohort | Case | |
|---|---|---|
| N=25 (%) | ||
| Age at systemic JIA diagnosis, mean±SD (range) years | 7.4±5.5 (1.1–17.2) | 2.7 |
| Disease duration, mean±SD (range) months | 51.6±48.8 (8–173) | 37 |
| Female | 19 (76) | Yes |
| Race | ||
| White | 17 (68) | Yes |
| African American | 7(28) | |
| Asian | 1 (4) | |
| Other/unknown | 0 (0) | |
| Ethnicity | ||
| Hispanic | 5 (20) | |
| Non-Hispanic | 20 (80) | Yes |
| JIA symptoms (ever had during disease course) | ||
| Fever | 25 (100) | Yes |
| Rash | 23 (92) | Yes |
| Arthritis | 25 (100) | Yes |
| Lymphadenopathy | 19 (76) | Yes |
| Hepatomegaly/splenomegaly | 20 (80) | Yes |
| Serositis | 14 (56) | No |
| Pulmonary symptoms | ||
| Age at start of pulmonary symptoms, mean±sd (range) years | 11.7±5.2 (3.5–18.8) | 6 |
| Disease duration at pulmonary diagnosis, mean±sd (range) months | 50.6±44.6 (8–160) | 37 |
| Time between pulmonary symptoms to diagnosis, mean±sd (range) months | 3.1±3.2 (0–10) | 4 |
| Time between pulmonary diagnosis to death (n=17), mean±sd (range) months | 10.2±13 (0–44) | 0 |
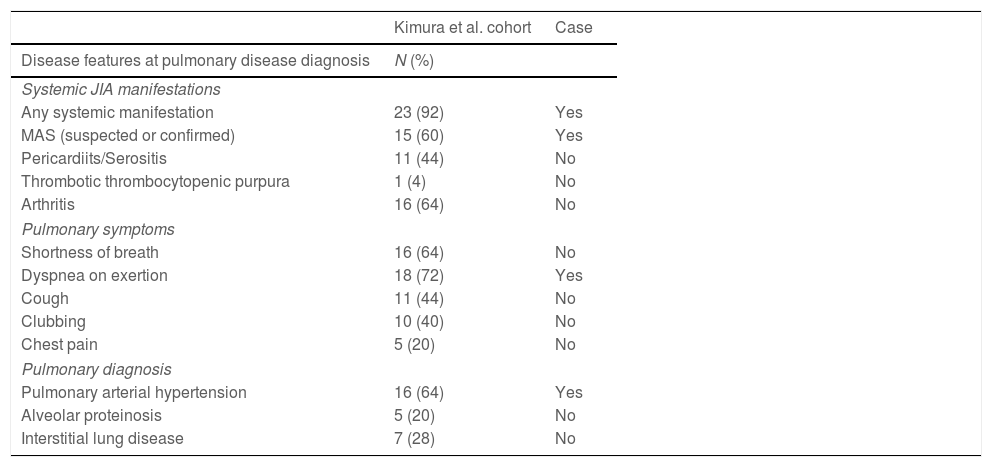
Clinical features at pulmonary disease diagnosis in Kimura et al. cohort and the patient case. Also, treatments being taken at the time of development of pulmonary symptoms or discontinued within a month prior to symptoms in the Kimura et al. cohort and in the patient case.
| Kimura et al. cohort | Case | |
|---|---|---|
| Disease features at pulmonary disease diagnosis | N (%) | |
| Systemic JIA manifestations | ||
| Any systemic manifestation | 23 (92) | Yes |
| MAS (suspected or confirmed) | 15 (60) | Yes |
| Pericardiits/Serositis | 11 (44) | No |
| Thrombotic thrombocytopenic purpura | 1 (4) | No |
| Arthritis | 16 (64) | No |
| Pulmonary symptoms | ||
| Shortness of breath | 16 (64) | No |
| Dyspnea on exertion | 18 (72) | Yes |
| Cough | 11 (44) | No |
| Clubbing | 10 (40) | No |
| Chest pain | 5 (20) | No |
| Pulmonary diagnosis | ||
| Pulmonary arterial hypertension | 16 (64) | Yes |
| Alveolar proteinosis | 5 (20) | No |
| Interstitial lung disease | 7 (28) | No |
| Treatments | Number of patients (%) | Exposure, months±DS | Case, yes/no treatment and exposure time (months) |
|---|---|---|---|
| Corticosteroids | 24 (96) | 47.3±48.2 (3–161) | Yes (4) |
| Cyclosporine | 7 (28) | 6.3±7.3 (1–22) | No |
| Etoposide | 1 (4) | 1 | No |
| Gold | 1 (4) | 53 | No |
| Methotrexate | 13 (52) | 32.9±(1–126) | Yes (35) |
| Thalidomide | 1 (4) | 22 | No |
| IL-1 inhibitor (any) | 12 (48) | 15.1±155.0 (3–47) | |
| Anakinra | 10 (40) | 16.9±15.9 (3–47) | No |
| Canakinumab | 1 (4) | 6 | Yes (11) |
| Rilonacept | 1 (4) | 6 | No |
| TNF inhibitor (any) | 3 (12) | 17.0±13.1 (2–26) | |
| Adalimumab | 2 (8) | 12.5±14.9 (2–23 | No |
| Etanercept | 1 (4) | 26 | No |
| Tocilizumab | 2 (16) | 6.0±7.1 (1–11) | No |
The most common clinical signs and symptoms in sJIA patients with pulmonary disease did not differ from the ones usually seen in this disease.3–5 However, the fact that most patients had MAS during their disease course or at PC diagnosis suggests that they are patients with higher and more severe systemic activity. Our patient was younger than previously published children, but systemic manifestations were prominent and had suffered two MAS episodes.3
Dyspnoea and fatigue are the most frequent symptoms in children with pulmonary hypertension. Other manifestations as shortness of breath, cough or chest pain are less common. Dyspnoea was the only symptom referred by our patient and was attributed to disease activity. The time from the start of the sJIA symptoms to the PC diagnosis lower than published patients (Table 1).
Chest X-ray, ECG and echocardiogram are useful to suspect pulmonary hypertension with advantages of being cost-effective and accessible. However, cardiac catheterization remains the gold standard to diagnose and evaluate the severity and indication of drug therapy. A complete evaluation should also include a pulmonary function test, a chest computed tomography and cardio-pulmonary exercise testing.3–7 In our case, due to the age of our patient, the functional test might not have been useful. As described, the echocardiogram and a chest X-ray chest performed months before the death did not show cardiomegaly or suggested pulmonary hypertension.7
Despite significant progress in treatment during the last years, pulmonary hypertension has poor prognosis. Two-thirds of the sJIA patients described by Kimura et al. died within a mean of 8.8±11.36 months from diagnosis of their pulmonary disease. Only in another case, the diagnosis was made after the death of the patient. Calcium channel blockers, endothelin receptor antagonists, phosphodiesterase 5 inhibitors and prostacyclins, alone or in combination, are used to achieve symptomatic control to improve the quality of life. In children with progressive and severe pulmonary hypertension, atrial septosomy or lung transplant can be considered.8,9
In summary, we present a case of a patient with sJIA that died as a consequence of pulmonary artery hypertension secondary to pulmonary veno-occlusive disease. High disease activity and the use of multiple therapies including biologic DMARD, should act as a red flag to the clinicians to consider PC. The combination of chest X-ray, ECG and echocardiogram are the best tests to make an early diagnosis. The main objective is to initiate symptomatic therapies, to anticipate instabilities and to improve the quality of life of these children. More studies and case description are needed to increase the knowledge about this uncommon complication and to improve management.
Conflict of interestNone.


