




La artritis reumatoide (AR) es una enfermedad crónica e incapacitante que afecta a individuos en etapas productivas de la vida. El tratamiento moderno de la AR incluye la denominada terapia “biológica” basada en proteínas recombinantes, modificadoras de procesos biológicos, con efectos terapéuticos potentes y diferentes mecanismos de acción, pese a lo cual persisten fracasos terapéuticos.
Un tratamiento que prevenga la discapacidad en AR debe instituirse en forma temprana, antes del desarrollo de secuelas, e idealmente con mínima posibilidad de fracaso terapéutico. No existen criterios clínicos o de laboratorio que identifiquen a pacientes con mayor probabilidad de respuesta a distintas formas de terapia, lo que retarda el control de la AR y afecta a la prevención de discapacidad. El estudio de la diversidad genética, por medio de polimorfismos de una sola base (SNP) con sistemas de microarreglos (MA), permite el análisis detallado de los factores genéticos asociados a una enfermedad, lo cual empieza a utilizarse en AR. Los polimorfismos con mayor asociación con AR ocurren primordialmente en genes que codifican proteínas relacionadas con el inicio de la respuesta inmunitaria y/o con el control de la actividad celular, además de genes relacionados con la reparación tisular. El significado específico de esto apenas empieza a estudiarse. Por otro lado, la proteómica estudia los perfiles de expresión proteínica en cualquier individuo a múltiples niveles.
Ambos tipos de estudios ayudarían a conocer los patrones de expresión génica en AR comparados con la población general. Además de ayudar a conocer la patogenia de la AR, los perfiles proteómicos y genómicos pueden utilizarse para diseñar sondas que identifiquen a individuos con riesgo de desarrollar AR, predigan en forma individualizada la respuesta a distintos esquemas terapéuticos y que permitan seguir la respuesta biológica a la terapia.
Rheumatoid arthritis (RA) is a chronic, disabbling disease that affects individuals during the productive years of their lives. Modern treatment for RA includes the so called “biologic” therapy, which is based on recombinant proteins that modify the biologic processes. These agents have potent therapeutic effects and different mechanisms of action. Nevertheless, therapeutic failure still prevails. Treatment that prevents disability in RA must be started in an early manner, before the development of complications and, ideally, with a minimum possibility of therapeutic failure. As yet, there are no clinical or laboratory criteria to identify those patients with a higher probability of responding to particular types of therapy, delaying control of RA ad affecting the prevention of incapacity.
Research into gene diversity through single-nucleotide polymorphisms (SNPs) by means of microarray systems, allows the detailed analysis of gene factors associated to a given disease. SNPs have been recently applied to the study of RA, where the major polymorphisms associated to RA occur primarily in genes that code for proteins related to the initiation of an immune response and/or the control of cellular activity in the immune system, in addition to genes related to tissue repair.
The specific meaning of these findings is in its initial stages of research. On the other hand, proteomics relate to the analysis of protein expression profiles at multiple levels. Both types of studies will contribute to the knowledge of patterns of gene expression in RA compared to the general population, and will allow an understanding of the pathogenesis of RA. Moreover, proteomic and genomic profiles can be employed to designs probes that identify individuals with the risk of developing RA, individually predict the response to different therapeutic modalities (pharmacogenomics) and for the follow-up of the biologic response to therapy.
Pese al gran avance terapéutico que ha significado la introducción de los llamados agentes biológicos en el tratamiento de la artritis reumatoide (AR), no debemos aún sentirnos satisfechos, ya que probablemente no estamos ni a la mitad del camino y lo logrado hasta ahora, aunque bueno, es incompleto y el tratamiento sigue siendo, en gran parte, empírico. Esta revisión tiene por objeto actualizar en forma crítica el conocimiento de la AR, en especial su patogenia molecular, para entender las bases de la terapia actual y buscar nuevas terapias más eficaces, dirigidas a posibles subgrupos de AR. En la primera parte se hace un resumen del estado actual del conocimiento de la terapia de la AR, seguido de una revisión de la genética de la AR, y, finalmente, un ensayo del posible papel de los distintos genes implicados en la susceptibilidad a la AR en su patogenia para abrir el campo para la propuesta de nuevos blancos terapéuticos.
Estado actual del conocimiento y tratamiento de la ARParece redundante afirmar que la AR es la afección inflamatoria de más impacto en la reumatología. Si su prevalencia es del 0,3 al 0,5%1–6, considerando adultos de 20 a 64 años de edad (54,5 millones según el INEGI-2005), el número estimado de pacientes con AR en México sería de 169.000 a 273.000, mientras que en España, el padrón de 2006 reveló una población de 44,7 millones de habitantes, con 28,6 millones de adultos de 20–64 años de edad (INE); considerando una prevalencia del 0,5% en adultos2,7, serían 143.319 casos. En general, se acepta que prácticamente todos los pacientes con AR referidos al reumatólogo requieren tratamiento con fármacos antirreumáticos modificadores de la enfermedad (FARME), aunque éstos podrían no representar a todos los casos con AR.
Ante la dificultad para prevenir una afección de etiología oscura, el objetivo del tratamiento de la AR sigue siendo inducir remisión de la actividad, actualmente inalcanzable en la mayoría de los casos. Así, la alternativa es obtener el mejor control posible de la actividad, alivio sintomático, recuperar la calidad de vida y la capacidad funcional para actividades cotidianas y laborales, así como prevenir la mortalidad. Para esto es necesario detener o retrasar el daño estructural, lo que se logra modificando el proceso biológico de daño tisular de la AR.
Los FARME son agentes capaces de modificar la biología y el pronóstico de la AR, de los cuales el metotrexato (MTX) en dosis de hasta 20mg semanales sigue siendo el tratamiento estándar, tanto que se usa también como fármaco de base para acompañar el tratamiento con agentes biológicos8–16. Los otros FARME que han mostrado utilidad en el tratamiento de la AR son sulfasalazina y cloroquina, sobre todo combinadas con MTX, y la leflunomida con o sin MTX. Las sales de oro y la D-penicilamina cada vez se utilizan menos. Un denominador común de estos medicamentos es que se desconocen las bases de su efecto terapéutico en la AR, pese a conocerse sus mecanismos farmacológicos, excepto la ciclosporina A y el FK50617–19, los cuales actúan inhibiendo dos moléculas distintas necesarias para la activación de la calcineurina, que es una fosfatasa necesaria para la activación del factor nuclear de linfocitos T (NFAT), indispensable para la activación celular. Un posible FARME adicional es la rapamicina, que inhibe la activación celular a través de otra vía, pero que no se ha evaluado sistemáticamente en AR (fig. 1).
 – desfosforilación (P–). En rojo, por marcación de degradación por ubicuitina (Ub+). Los sitios de acción de abatacept (Ab), ciclosporina (Cy) y rapamicina (Rap).")
Vías de transducción de señales en linfocitos T. Las flechas de punta redonda indican inhibición, en azul por fosforilación (P+) – desfosforilación (P–). En rojo, por marcación de degradación por ubicuitina (Ub+). Los sitios de acción de abatacept (Ab), ciclosporina (Cy) y rapamicina (Rap).
Entre los FARME biológicos, la terapia con anticuerpos anti-TNF fue la primera con éxito terapéutico, después de intentos fallidos con anticuerpos anti-CD4, anti-ICAM1 y otros más, que no pasaron de las primeras fases experimentales, pese a que su uso se fundamentaba en las supuestas bases patogénicas de la AR. Es posible que el fracaso de estos tratamientos, especialmente anti-CD4, se haya debido a que se utilizaron en pacientes con AR avanzada y quizá nunca lleguemos a saber si su uso en AR temprana hubiera significado otra cosa. Además, es de extrema importancia fundamentar bien su uso para evitar eventos catastróficos, como la tormenta de citocinas que ocurrió en voluntarios sanos tratados con un anticuerpo agonista anti-CD2820.
Con todo, la terapia anti-TNF, que incluye el etanercept (receptor soluble de TNF) y los anticuerpos monoclonales anti-TNF infliximab y adalimumab, tiene una eficacia variable. Por ejemplo, con infliximab se obtuvo respuesta clínica del 51,8 frente al 17% en controles a 6 meses (p < 0,001), pero muy pocos tuvieron remisión15,21,22. Con etanercept en monoterapia (25mg 2 veces por semana), el 59% de los pacientes obtuvieron mejoría de ACR20 a los 6 meses, frente al 11% de los controles; el 40% mejoró en ACR50 y el 25%, en ACR70. Etanercept asociado a MTX es efectivo a corto y largo plazo23–29. Con adalimumab más MTX, el 62% de los pacientes alcanzaron un ACR50 al año frente al 46% en el grupo de MTX solo. A los 2 años, el 59% de los pacientes en terapia combinada lograron ACR50, comparados con el 43% del grupo de MTX27,30–32. Estos resultados, que sólo son ejemplos de una literatura muy vasta, muestran que, pese a ser efectivos, los anti-TNF rara vez inducen remisión. Un efecto notable de los anti-TNF es su capacidad de disminuir la progresión del daño estructural, evidenciado por un menor número de erosiones y menor pérdida del cartílago, aun en pacientes sin mejoría sintomática.
El efecto secundario más importante de los anti-TNF es el desarrollo de infección (casi siempre reinfección endógena) por Mycobacterium tuberculosis, que es su principal limitante (no contraindicación), pero que, como hallazgo un tanto inesperado, ayudó a conocer que una función del TNF es prevenir la diseminación de agentes intracelulares, posiblemente por ser indispensable para la formación de granulomas.
Rituximab es un anticuerpo contra la molécula CD20, expresada por linfocitos B, desde sus precursores en médula ósea hasta antes de células plasmáticas33,34. El rituximab, que elimina linfocitos B aparentemente induciendo su apoptosis, se ha utilizado con éxito en el tratamiento de linfomas no hodgkinianos de estirpe B35,36 y parece ser eficaz en un número relativamente bajo de pacientes con AR, en los que la mejoría puede ser notable, con el 13% de remisión después del segundo ciclo de administración10,37. Dado que el papel patogénico de los linfocitos B en AR no está claro (revisado en Díaz-González et al38), se desconocen las bases del efecto terapéutico del rituximab en la AR.
Un tercer agente biológico eficaz en AR es la proteína quimérica abatacept, formada por el receptor CTLA-4 (expresado por linfocitos T activados) y la fracción Fc de IgG1 humana. Abatacept funciona como antagonista al unirse a CD86 y CD80, inhibe su unión con su receptor CD2839–44, y así previene la activación de linfocitos T al inicio de la respuesta inmunitaria por inhibición competitiva45,46. En AR, la respuesta ACR20 a las 12 semanas es del 53%; ACR50, del 51%, y ACR70, del 28%; con abatacept más MTX, comparados con placebo y MTX, del 41, el 35 y el 5%, respectivamente.
En conclusión, todos los FARME biológicos disponibles son eficaces y suprimen la actividad, y aunque clínicamente no parecen muy superiores a MTX solo o combinado con cloroquina y/o sulfasalazina47–49, los biológicos retardan la progresión de las lesiones radiológicas en AR resistente a tratamiento convencional. No obstante, hay muchos pacientes que no responden a uno o más agentes biológicos, especialmente si éxito se define como inducción de remisión.
El hecho de que no todos los pacientes respondan al mismo tratamiento indica que la patogenia de la AR, como enfermedad compleja, es heterogénea, lo cual concuerda con la variedad de factores genéticos asociados a ella. La respuesta a uno u otro tratamiento podría depender del mosaico genético de cada paciente, que refleja diferencias patogénicas entre ellos. Actualmente no sabemos si un paciente con resistencia a un FARME biológico podría responder a otro o incluso a combinaciones. Es esencial contar con tratamientos racionales individualizados, dirigidos a puntos cruciales de la patogenia molecular de la AR.
La importancia de conocer la genética de la ARAunque muchos aspectos de su patogenia siguen siendo oscuros, el origen de la inflamación en la AR es de tipo autoinmune. Un cúmulo de evidencias64–69,77 apoya la participación de factores genéticos en la susceptibilidad a AR, que interactúan con factores ambientales para el desarrollo de la enfermedad. La AR puede presentarse como casos múltiples en una misma familia y la susceptibilidad a desarrollarla se hereda en forma multigénica, aunque el significado directo de cada uno de los polimorfismos asociados, incluso los más estudiados, aún no se comprende completamente.
Por ejemplo, algunas variantes genéticas asociadas a AR ocurren en la parte del gen que codifica directamente la proteína. En estos casos, lo más probable es que el mecanismo implicado sea una función alterada de la proteína, ya sea pérdida o ganancia. Sin embargo, otras variantes ocurren en regiones no codificantes del genoma, las cuales pueden ser de varios tipos:
- 1.
Polimorfismos dentro del mismo gen:
- –
En regiones reguladoras (promotores y/o enhancers). Pueden afectar a la tasa de transcripción y, consecuentemente, la abundancia de la proteína.
- –
En intrones. Mecanismo de asociación poco claro, podría afectar al uso de un exón (splicing) o enhancers intrónicos.
- –
Al final del gen (región 3' no traducida). Podrían afectar a la estabilidad del ARN mensajero y, consecuentemente, la abundancia de la proteína.
- –
- 2.
Polimorfismos fuera del gen (5' o 3'). Podrían afectar a la estructura de la cromatina, con implicaciones en la expresión del gen, incluso pueden suprimirse.
- 3.
Polimorfismos dentro o fuera del gen. Especialmente en regiones no codificantes o en el mal llamado ADN “basura” que afectaran a la secuencia y, consecuentemente, la función de algún micro-ARN regulador de la expresión de otros genes.
Independientemente del mecanismo, un gen afectado podría no estar implicado directamente con un mecanismo patogénico, sino que podría ser un gen regulador de la expresión de los genes causales de un mecanismo patogénico dado. Mientras la genómica funcional no se defina a fondo, el conocimiento preciso de lo anterior es imposible. Así, describiremos algunas de las asociaciones genéticas de la AR y se discutirá el posible efecto de los polimorfismos descritos.
Es bien sabido que el principal factor genético asociado al riesgo de desarrollar AR, ampliamente confirmado en distintos grupos étnicos, es un polimorfismo en el locus DRB150–55, perteneciente a la región clase II del complejo principal de histocompatibilidad (MHC), que codifica la cadena beta de la molécula HLA-DR. Este gen tiene más de 500 alelos; los que codifican la secuencia de aminoácidos Gln-Lys-Arg-Ala-Ala (QKRAA, código internacional de una letra) en los residuos 70–74 de HLA-DRβ1 (epítopo compartido) se asocian a AR. De las enfermedades autoinmunitarias, sólo la diabetes mellitus tipo 1 se asocia a un riesgo mayor con polimorfismos en este locus (p = 2,44 × 10-134). Subsecuentemente se han encontrado otros genes asociados a AR, algunos en todos los grupos étnicos, mientras que otros varían dependiendo de las poblaciones estudiadas.
Hasta hace poco, los estudios genéticos en enfermedades complejas, como AR, se limitaban a uno o unos cuantos genes por grupo de investigadores. La secuenciación del genoma humano y la disponibilidad de métodos masivos de genotipificación, junto con metodología estadística avanzada, han hecho posible estudiar simultáneamente los patrones de variación genética en todo el genoma en poblaciones de individuos.
Los polimorfismos de un solo nucleótido (SNP, por sus siglas en inglés single nucleotide polymorphism) son variaciones que en la población general ocurren cada 100–300 bases en la secuencia del ADN del genoma, tanto en regiones codificantes como no codificantes, y representan el 90% de la variabilidad genética humana56–59. Algunos SNP tienen un gran impacto en biología y respuesta a agentes ambientales, incluidos virus, bacterias, toxinas y medicamentos. Estudios iniciales de SNP aislados identificaron polimorfismos asociados a AR, que parecen ser compatibles con ciertos aspectos de su patogenia.
Hoy en día, la genotipificación puede hacerse con sistemas de microarreglos de SNP a gran escala (SNPGE), que identifican hasta más de 500.000 SNP en todo el genoma, codificante o no. Los estudios de SNPGE analizados mediante el Proyecto Internacional de Mapeo de Haplotipos (HapMap)60, que son bases de datos públicas que contienen más de dos millones de SNP con frecuencias alélicas conocidas y verificadas, están ya contribuyendo a la identificación de las variantes genéticas que predisponen a enfermedades complejas, como la AR61, algunas de las cuales podrían variar en distintos grupos étnicos. Lo anterior, en conjunto con estudios de expresión génica, por genómica y/o proteómica, contribuirá significativamente a definir la patogenia de la AR. Dos estudios recientes de SNPGE, uno en 2.000 pacientes británicos caucásicos con AR y 3.000 controles sanos61 y el otro en norteamericanos y suecos62, confirmaron que DRB1 es el principal locus de susceptibilidad a AR (p = 3,44 × 10-76 y p < 1 × 10-100, respectivamente) y que el segundo locus de susceptibilidad es el gen de la fosfatasa de tirosina PTPN22 (p = 4,9 × 10-26 y p = 2 × 10-11, respectivamente), que también se asocia a otras enfermedades autoinmunitarias como lupus eritematoso sistémico (LES) y diabetes tipo 161,63,64.
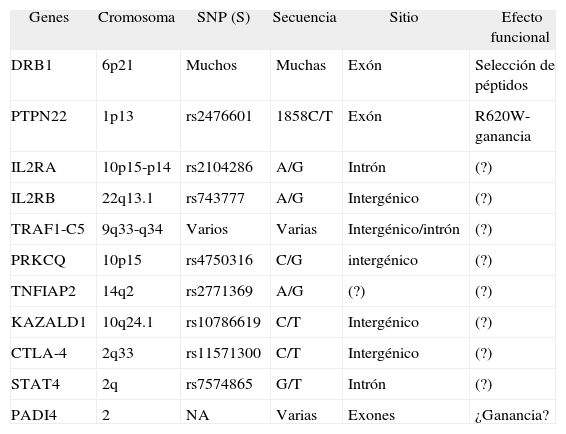
Otros polimorfismos, previamente descritos en AR, se confirmaron sólo en uno u otro de los estudios de SNPGE (tabla 1). En el estudio británico hubo asociación con los genes que codifican las cadenas alfa y beta del receptor de interleucina (IL)-2 (IL-2α, IL-2β; ambas, p = 10-5), y en los genes del inhibidor de TNF (TNFAIP2), granzima B (GZMB), proteincinasa C-0 (PKCθ) y el inhibidor de proteasas KAZALD1; todos con p = 10-4-10-5. En el estudio norteamericano/sueco no se encontró ningún SNP en estos genes, pero sí en una región de 100kb del cromosoma 9, donde se encuentran los genes TRAF1 y de la fracción C5 del complemento (p = 2,8 × 10-8), además de una asociación positiva, pero menor en los genes del receptor CD40 (p = 3 × 10-6), el receptor 1 de bradicinina (BDKR1, p = 1 × 10-5), un grupo de genes del cromosoma 17 que codifican las quimiocinas CCL1, CCL3 y CCL8 (4 × 10-5) y con un SNP en un intrón del gen que codifica la molécula STAT4 (cromosoma 2), de la vía de señalización por citocinas. Cualquier alteración en estos genes podría participar en el desarrollo de autoinmunidad (tabla 1). Un SNP de interés potencial, asociado a AR en asiáticos, es en el gen de la peptidilarginina deiminasa (PADI) 465,66. Esta familia de enzimas hidroliza grupos imino de arginina a hidroxilo en proteínas transformándolas en citrulina y producen las proteínas citrulinadas presentes en el organismo (véase más adelante). Otros polimorfismos asociados a AR, también en japoneses, son en los genes NFKBIL1, SLC22A4 y RUNX167.
Principales polimorfismos en artritis reumatoide
| Genes | Cromosoma | SNP (S) | Secuencia | Sitio | Efecto funcional |
| DRB1 | 6p21 | Muchos | Muchas | Exón | Selección de péptidos |
| PTPN22 | 1p13 | rs2476601 | 1858C/T | Exón | R620W-ganancia |
| IL2RA | 10p15-p14 | rs2104286 | A/G | Intrón | (?) |
| IL2RB | 22q13.1 | rs743777 | A/G | Intergénico | (?) |
| TRAF1-C5 | 9q33-q34 | Varios | Varias | Intergénico/intrón | (?) |
| PRKCQ | 10p15 | rs4750316 | C/G | intergénico | (?) |
| TNFIAP2 | 14q2 | rs2771369 | A/G | (?) | (?) |
| KAZALD1 | 10q24.1 | rs10786619 | C/T | Intergénico | (?) |
| CTLA-4 | 2q33 | rs11571300 | C/T | Intergénico | (?) |
| STAT4 | 2q | rs7574865 | G/T | Intrón | (?) |
| PADI4 | 2 | NA | Varias | Exones | ¿Ganancia? |
Véase el texto para detalles de las abreviaturas.
Los SNP están referidos de acuerdo con su nomenclatura en NCBI y en Affymetrix.
De todo lo anterior podemos concluir lo siguiente:
- –
El locus principal de asociación a AR (HLA-DRB) codifica una de las proteínas más importantes en el inicio de la respuesta inmunitaria adaptativa.
- –
La mayoría de los polimorfismos adicionales asociados a AR ocurren en genes que codifican proteínas que participan en la regulación de la respuesta inmunitaria y/o el proceso inflamatorio.
- –
Algunos polimorfismos asociados a AR ocurren en genes diversos que afectan a la estructura antigénica de las proteínas (como la citrulinación), procesos de reparación tisular (como KAZALD1) y posiblemente otros que afectan la respuesta del organismo a la autoagresión inmunológica.
- –
De todos los polimorfismos descritos, sólo 2 (DRB1 y PTPN22) afectan a la secuencia de la proteína, todos los demás ocurren en regiones no codificantes del ADN.
- –
Algunos polimorfismos asociados a AR varían en distintos grupos étnicos.
Todo indica que la AR es un proceso autoinmunitario dependiente de linfocitos CD4+ que inducen inflamación sinovial crónica; entre otras evidencias, las respuestas a ciclosporina A17,18, FK50619 y abatacept39–44 apoyan esta aseveración. Estos estimulan macrófagos y fibroblastos para producir citocinas como IL-1, IL-6 y TNF68, además de proteasas, que degradan cartílago y hueso subcondral. Sin embargo, las causas subyacentes que llevan a la activación de estos sistemas efectores son desconocidas. Así, en plena era de la genómica y la proteómica, la etiología de la AR sigue siendo desconocida y el conocimiento de su patogenia sigue siendo superficial. La elaboración de un modelo que explique la patogenia de la AR debe tomar en cuenta, además de los hallazgos biológicos, las funciones de los genes con polimorfismos asociados a AR y los mecanismos de acción de algunos de los tratamientos con eficacia probada, pero sólo los medicamentos con mecanismos de acción conocidos.
Papel del epítopo compartidoEl epítopo compartido se refiere a la secuencia de aminoácidos QKRAA (Gln-Lys-Arg-Ala-Ala) en los residuos 70–74 de la tercera región polimórfica de la cadena HLA-DRβ1. Esta cadena es parte del dímero HLA-DR, que es uno de los isotipos de las MHCII humanas. Es bien sabido que la función de las MHCII69 es presentar péptidos derivados de proteínas del medio extracelular a los linfocitos T CD4+. Los péptidos presentados por las MHCII deben unirse a ellas y la secuencia 70–74 de HLA-DRβ se encuentra en el sitio de unión de los péptidos. Distintos aminoácidos en la secuencia 70–74 tienen como resultado la capacidad de unir péptidos diferentes; por lo tanto, la diferencia principal entre los individuos poseedores del epítopo compartido y los que no lo poseen es en el tipo de péptidos presentados por su molécula HLADR.
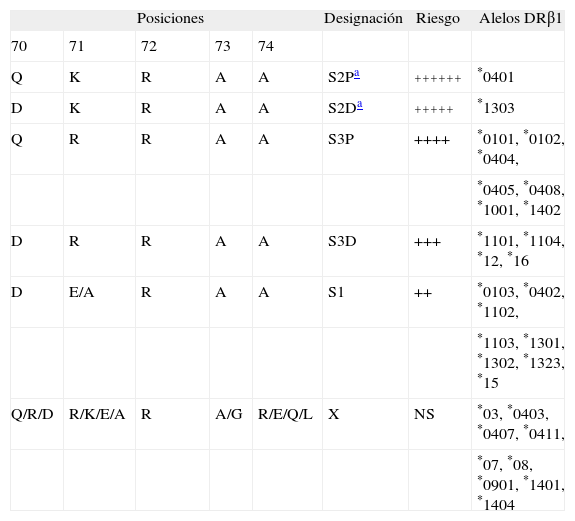
Recientemente se analizó en detalle la secuencia del epítopo compartido52,53, y se encontró que la susceptibilidad depende estrictamente de la secuencia RAA (arginina-alanina-alanina) en los aminoácidos 72–74, mientras que los residuos K o R (lisina o arginina) en posición 71 y, en menor grado, el residuo Q (glutamina) en la posición 70, además de contribuir al riesgo, se relacionan con la producción de autoanticuerpos y aparentemente con severidad de la AR (tabla 2). Lo notable de esto es que la arginina (R) dentro de esta secuencia tiene carga positiva, mientras que la alanina es neutra. Además, la lisina o la arginina en la posición 71, ambas aminoácidos básicos, aumentan la carga positiva total en esta región, mientras que la glutamina en posición 70 es un aminoácido polar, neutro. Así, la carga del epítopo compartido es firmemente positiva, lo que predice que los péptidos que se unan a esta molécula HLA-DR deben tener carga negativa (es decir, aminoácidos “ácidos”, como ácidos aspártico y glutámico) y no tener aminoácidos con carga positiva (lisina, arginina e histidina).
Riesgo de artritis reumatoide según las secuencias de DRβ1
| Posiciones | Designación | Riesgo | Alelos DRβ1 | ||||
| 70 | 71 | 72 | 73 | 74 | |||
| Q | K | R | A | A | S2Pa | ++++++ | *0401 |
| D | K | R | A | A | S2Da | +++++ | *1303 |
| Q | R | R | A | A | S3P | ++++ | *0101, *0102, *0404, |
| *0405, *0408, *1001, *1402 | |||||||
| D | R | R | A | A | S3D | +++ | *1101, *1104, *12, *16 |
| D | E/A | R | A | A | S1 | ++ | *0103, *0402, *1102, |
| *1103, *1301, *1302, *1323, *15 | |||||||
| Q/R/D | R/K/E/A | R | A/G | R/E/Q/L | X | NS | *03, *0403, *0407, *0411, |
| *07, *08, *0901, *1401, *1404 |
A: alanina; D: ácido aspártico; E: ácido glutámico; K: lisina; L: leucina; Q: glutamina; R: arginina.
Los grupos de susceptibilidad (S) se dividieron en 5 subgrupos de acuerdo con la secuencia de aminoácidos en los residuos 70–74 de DRβ1 que corresponden al riesgo de desarrollar AR y cursar con autoanticuerpos.
Existen alelos adicionales que no han sido clasificados en estas secuencias, pero que son mucho menos frecuentes en la población.
Lo anterior se ejemplifica considerando las secuencias de aminoácidos 70–74 en los alelos no asociados a AR, que tienen ácido aspártico (D) en posición 70 o ácido glutámico (E) en posición 71, ambos con carga negativa que contrarresta la carga positiva de la arginina 72. Además, en todos los alelos no asociados, los residuos en posición 74 (arginina/glutámico/glutamina/leucina, R/E/Q/L) tienen características fisicoquímicas muy distintas de la alanina. Así, el posible resultado neto de todo esto es que los alelos asociados a AR unirían péptidos con carga menos positiva, con implicaciones importantes en el blanco de la respuesta inmunitaria que se comenta en detalle más adelante.
Linfocitos B, proteínas citrulinadas y anticuerpos contra ellasDe los autoanticuerpos característicos de AR, los que aparecen más temprano son los que reconocen péptidos citrulinados cíclicos (CCP) y/o fibrinógeno citrulinado70,71. La citrulina es precursor de la biosíntesis de arginina, sintetizada a partir de ornitina, con adición de amonio y CO2. Por otro lado, la NO sintetasa hidroliza la arginina a citrulina y NO.
La citrulina no tiene codones ni ARN de transferencia para su incorporación en proteínas. Por lo tanto, las proteínas citrulinadas son producto de la hidrólisis del grupo imino de los residuos de arginina en proteínas por las PADI, que tienen varias isoformas y diferentes patrones de expresión tisular. Como se mencionó, el gen de PADI4 tiene un SNP que se asocia a AR en asiáticos66,72.
El grupo imino de la arginina tiene carga positiva, que en la citrulina es un hidroxilo, con carga neutra. Así, la hidrólisis de citrulina a arginina neutraliza su carga positiva en las proteínas. Por lo tanto, los péptidos citrulinados tienen carga menos positiva que la misma secuencia con arginina, lo cual necesariamente afecta a su unión a la molécula MHCII (como HLA-DR). Como ya se ha mencionado, la secuencia 70–74 de DRβ1 se encuentra en un sitio crucial para la unión de péptidos a HLA-DR, que en los individuos portadores de alelos con el epítopo compartido tiene carga positiva, favoreciendo la unión de péptidos con carga negativa o neutros (como los péptidos citrulinados) que así serían presentados a linfocitos T autorreactivos. Algunos estudios han confirmado que en pacientes con AR portadores del epítopo compartido hay un aumento en la respuesta de linfocitos T a péptidos citrulinados52,65,71.
Aunque se desconocen las consecuencias funcionales del polimorfismo de PADI4, éste podría afectar positiva o negativamente a la capacidad de citrulinación proteínica y posiblemente la inmunogenicidad de algunas proteínas; por un lado, para su presentación a linfocitos T por las moléculas HLA-DR con epítopo compartido y, por otro, para la especificidad de los anticuerpos anti-CCP. En concreto, los linfocitos B específicos contra proteínas citrulinadas las captarían por medio de su receptor (inmunoglobulina de superficie o BCR). A esto seguiría su endocitosis y degradación parcial (procesamiento) a péptidos (también citrulinados), algunos de los cuales podrían unirse a la molécula HLA-DR y ser presentados a los linfocitos T autorreactivos, lo cual es necesario, aunque claramente insuficiente para el inicio de la autoinmunidad. La mayoría de los individuos portadores del epítopo compartido no tienen AR, a menos que sean también portadores de otros genes de susceptibilidad en un mosaico de combinaciones aún no predecible.
Genes que codifican proteínas de activación y de regulación celular. Héroes y villanosEs importante ser muy cautos en la interpretación de los datos genéticos, ya que siempre deben ser confirmados y su significado es difícil de definir. No obstante, algunas asociaciones parecen genuinas y la información funcional en ellas indica implicaciones patogénicas potencialmente importantes.
De los genes de susceptibilidad a AR, muchos codifican proteínas que participan en la activación o inhibición de funciones celulares, especialmente en linfocitos T. El segundo gen de susceptibilidad para AR, la fosfatasa de tirosina PTPN22, regula negativamente la activación de linfocitos T. Los dos alelos más frecuentes de PTPN22 son PTPN22-1858C y 1858T (sustitución única en el nucleótido 1858, que resulta en diferencia de un aminoácido en la posición 620; arginina para 1858C (PTPN22-620R) y triptófano para 1858T (PTPN22-620W). Las principales proteínas desfosforiladas por PTPN22 en linfocitos T son las cinasas de tirosina (PTK) Lck y ZAP7073. Ambas PTK tienen dos sitios principales de fosforilación, uno que inhibe su función enzimática y otro que la activa. Así, Lck 505P es inactiva, mientras que Lck 394P es activa. Asimismo, ZAP70-319P es inactiva y ZAP70-493P es activa. PTPN22 desfosforila las formas activas: Lck 505P y ZAP70 493P, por lo que su efecto es una inhibición de la función de estas PTK y, consecuentemente, de la activación de linfocitos T a través del TCR (figs. 1 y 2).

Selección negativa de linfocitos T y B autorreactivos de acuerdo con su umbral de activación. Izquierda, lo que ocurre en individuos sanos. Centro, exceso de clones autorreactivos por un umbral alto de activación que lleva a persistencia de clones autorreactivos que están sobrerrepresentados en individuos susceptibles portadores de PTPN22 620W. Derecha, situación hipotética en individuos con umbral bajo de activación, con clones autorreactivos disminuidos pero capacidad de activación aumentada.
Inesperadamente, un estudio reciente encontró que el alelo de PTPN22 asociado a AR, PTPN22-620W tiene mayor actividad catalítica que 620R74; es decir, inhibe más eficientemente la señalización del TCR. Así, su umbral de activación en individuos portadores de PTPN22-620W es más alto, por lo que los pacientes con enfermedades autoinmunitarias tendrían, paradójicamente, una respuesta inmunitaria de menor intensidad. Entonces, el posible papel de este cambio de un aminoácido en PTPN22 podría explicarse en dos formas alternas, pero no excluyentes. Por un lado, los linfocitos T de pacientes con AR tendrían una menor capacidad de respuesta a antígeno; sin embargo, durante la ontogenia, esto afectaría a la eficiencia para eliminar clones autorreactivos. Así, aunque en AR el umbral de activación de linfocitos T sería alto, el número de clones autorreactivos estaría elevado (fig. 2). Alternativamente, en la periferia, en los portadores del alelo de susceptibilidad la inducción de linfocitos T reguladores podría ser menos eficiente. La primera posibilidad es compatible con un modelo de artritis espontánea, semejante a AR, en ratones, en los que una mutación en ZAP70, que disminuye su capacidad de señalización (similar a lo que ocurre en los individuos con PTPN 620W), disminuye la deleción clonal en el timo, por lo que los linfocitos T autorreactivos pasan a la periferia y, eventualmente, causan artritis autoinmunitaria75.
Otros polimorfismos asociados a AR son en los genes que codifican las cadenas alfa y beta del IL-2R, en ambos casos, en un intrón61. Este receptor recibe señales de una de las principales citocinas de linfocitos T (IL-2). Al inicio de la respuesta inmunitaria, la IL-2 es indispensable para la proliferación de linfocitos T y B; sin embargo, una vez activados, la IL-2 induce muerte por activación, por lo que es necesaria para que el curso de una respuesta inmunitaria sea autolimitado. Además, la IL-2 es esencial para la diferenciación de un subgrupo de los linfocitos reguladores (Treg), que moderan la respuesta inmunitaria76. La ausencia congénita de IL2Rβ y/o IL-2Rα en modelos experimentales conlleva el desarrollo de afecciones autoinmunitarias77.
Otros polimorfismos asociados a AR ocurren en genes que codifican inhibidores del proceso inflamatorio, como TNFAIP2 y NFKBIL1, en intrones78,79. Mientras que otro polimorfismo es cercano al gen que codifica la PKCθ61, que es la principal cinasa de serina y treonina que transduce señales positivas del receptor de linfocitos T (TCR). Los polimorfismos en la región TRAF1-C5 son importantes, ya que TRAF1 participa en la señalización del TNF y la ausencia de C5 en ratones los hace resistentes al desarrollo experimental de artritis. Finalmente, otros polimorfismos con significado desconocido son en los genes SLC22A4 y RUNX167, presentes también en otras enfermedades autoinmunitarias.
Las combinaciones de pérdida de algunos de estos genes y ganancia de otros, junto con el epítopo compartido, podrían explicar la patogenia de la autoinmunidad de la AR. Además, distintas combinaciones de estos polimorfismos podrían dar lugar a subgrupos de AR que podrían variar en su comportamiento clínico y/o en su respuesta a distintos tipos de terapia.
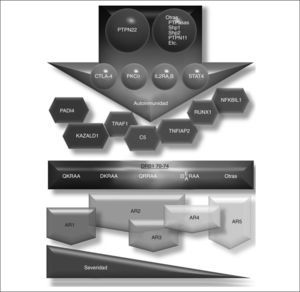
En resumen, para la patogenia de la AR, el hecho de que el principal gen de susceptibilidad a la AR (DRB1) sea una molécula MHCII tiene implicaciones importantes. La función de las MHCII es presentar péptidos a linfocitos T CD4+, que son los iniciadores de la respuesta inmunitaria adaptativa69 (véase más adelante). Sí los linfocitos T de pacientes con AR tienen un umbral alto de señalización a través de su TCR por efecto de PTPN22 (desfosforilación) o por señalización defectuosa (PKCθ), el resultado sería una eliminación clonal fallida, con aumento de clones autorreactivos. Si, por otro lado, hay un aumento de proteínas citrulinadas (polimorfismo en PADI4), captadas por linfocitos B y presentadas por DRB1 con la secuencia QKRAA, que presentan con mayor facilidad los péptidos citrulinados, se activarían los linfocitos T y B autorreactivos en forma recíproca. Estos últimos son los que causan la secreción de los anticuerpos anti-CCP, mientras que los primeros, por alguna razón aún oscura (véase posible explicación más adelante), migrarían al tejido sinovial.
Los defectos en linfocitos Treg, por errores en la señalización del TCR y/o IL-2R, con defectos en el control de la inflamación, por mayor señalización de TNF y otras citocinas inflamatorias (TNFAIP2 y NFKBIL1), harían que un estímulo antigénico leve resultara en una respuesta inflamatoria magnificada, que en individuos con defectos en la reparación tisular (KAZALD1) favorecería el desarrollo temprano de erosiones (AR de curso agresivo).
Toda esta secuencia hipotética de eventos se correlaciona con diversos hallazgos en pacientes con AR, algunos de los cuales no necesariamente tienen traducción genética directa, sino que muy probablemente sean efectos indirectos de los defectos en los genes mencionados. Los linfocitos T predominantes en el infiltrado sinovial de la AR son CD4+ y la principal citocina que secretan es IL-1768, que induce citocinas proinflamatorias, como TNF, IL-1 e IL-6, y quimiocinas, como IL-8, que atraen PMN. Los macrófagos activados en sinovial de AR causan daño tisular a través de TNF e IL-1, que inducen secreción de colagenasa y elastasa por sinoviocitos y condrocitos y degradan cartílago y hueso. La inflamación persistente con TNF induce neovascularización, hiperplasia sinovial, fibrosis (pannus); además, junto con RANK, activa osteoclastos, lo que lleva a la resorción ósea. La duración del proceso inflamatorio es proporcional a la destrucción de cartílago, hueso, tendones y ligamentos. La IL-6 induce proteínas de fase aguda, que son: pentraxinas (proteína C reactiva [PCR] y amiloide P [SAP]), complemento, fibrinógeno, además de anticuerpos y autoanticuerpos (como factor reumatoide y anticuerpos anti-CCP) por linfocitos B y células plasmáticas, favoreciendo el daño mediado por complejos inmunitarios. De hecho, la IL-6 es la causa directa o indirecta de muchas de las manifestaciones sistémicas de la AR.
Un aspecto que aún no está claro, pero que es necesario abordar para explicar la eficacia terapéutica del rituximab, es el papel de los linfocitos B en el proceso patológico de AR. Se ha propuesto que podría deberse a su función como célula presentadora de antígeno (CPA) a linfocitos T en la membrana sinovial. Debido a que en la sinovial (y en otros procesos inmunitarios) hay otras CPA, como las células dendríticas, con mejor capacidad para activar linfocitos T, el papel de los linfocitos B como CPA en AR sólo tendría sentido si los consideramos como una CPA particular presentadora de péptidos citrulinados y que la respuesta contra éstos, tanto de linfocitos T como B, tiene un papel preponderante en la AR (aunque no en todos los pacientes, ya que sólo alrededor del 25% responde a rituximab).
El posible mecanismo de acción del rituximab en AR podría ser la eliminación de linfocitos B productores de anticuerpos anti-CCP, si su papel en la patogenia de la AR es similar al de un modelo de artritis en ratones, en los que un autoanticuerpo contra una proteína ubicua80–82 transfiere la enfermedad pasivamente a ratones sanos. Pese a iniciarse con los autoanticuerpos, la artritis se establece cuando, secundariamente, la membrana sinovial es infiltrada por linfocitos T, macrófagos y otras CPA. Este proceso depende, además, de la participación de la fracción C5 del complemento, receptores Fcγ, TNF y células cebadas. Pese a haberse identificado todos estos factores como esenciales para la enfermedad, el papel preciso de cada uno de ellos aún no está claro.
Utilidad clínica de conocer las alteraciones moleculares de la ARSe conocen ya muchos de los mecanismos de daño estructural en AR y algunos de sus mediadores biológicos han sido plenamente identificados. Está claro que el pronóstico funcional de la AR depende en forma directa del control biológico de la inflamación, lo cual debe hacerse en el menor tiempo posible, ya que depende de dos variables principales: a) un diagnóstico temprano, y b) elegir la mejor opción terapéutica para cada paciente. Actualmente, ninguna de ellas es fácil de alcanzar.
El diagnóstico temprano de AR sigue siendo un problema; primero, porque no hay herramientas paraclínicas para establecer o descartar su diagnóstico, que se sigue basando en sus manifestaciones clínicas según los criterios del American College of Rheumatology (ACR), cuyo problema principal es que un aspecto esencial de casi todos sus incisos es la cronicidad (más de 6 semanas), además de que sólo se aplican a pacientes con enfermedad activa, ya que se basan en la observación directa de la inflamación por el médico.
El clínico avezado (cualquier reumatólogo con experiencia) puede presuponer, con un buen grado de certeza, que un paciente tiene AR aun antes de cumplir con los criterios del ACR. Aunque esto facilita establecer un tratamiento en forma más o menos temprana, no resuelve el problema de no poder predecir el mejor tratamiento para cada paciente de forma individual.
Por lo anterior, diversos grupos se han dedicado a definir lo que se denomina AR temprana, que es útil, pero no para estudios epidemiológicos, ya que sólo a los pacientes que cumplen los criterios del ACR se puede considerarlos como válidos; lo que deja fuera a los pacientes que, por su buena respuesta, no llegaron a acumular el número necesario de criterios para que se los considere con AR (pese a ser casos auténticos). La selección de pacientes con base en su positividad para anticuerpos anti-CCP es un avance sustancial con respecto a otros estudios paraclínicos. Sin embargo, aunque los anti-CCP están en la AR temprana, no todos los pacientes con AR los tienen y hay falsos positivos.
El componente de cronicidad de los criterios es otro aspecto que afecta a los estudios clinicoterapéuticos de AR, ya que en el momento de su inclusión en un estudio, y del inicio formal del tratamiento, un paciente con AR ha pasado más de 6 semanas con sinovitis, en que puede ocurrir daño estructural sustancial, pues la AR parece estar en forma subclínica por semanas o meses antes de ser detectable por exploración física.
Además de aportar al entendimiento de su patogenia, los perfiles genéticos y moleculares de la AR podrían utilizarse como marcadores pronósticos, de susceptibilidad, indicadores de respuesta o no a distintas terapias o bien ser la base para diseñar nuevos fármacos. Los estudios aislados de asociación genética son útiles, pero el número de genes que permiten estudiar es limitado. Una solución a ello es la genotipificación por microarreglos de SNP para identificar variantes genéticas asociadas a AR, los cuales pueden diferir entre distintos grupos étnicos. Ya nos hemos referido a dos estudios recientes de genotipificación masiva por medio de esta metodología en pacientes con AR.
Otra herramienta para conocer el perfil molecular de una enfermedad es la proteómica, que en lugar de genes estudia su producto final: las proteínas, las cuales reflejan el estado funcional de un organismo. En la célula, los genes se transcriben a ARN mensajero (ARNm) que codifica la secuencia de una o más proteínas por gen, dependiendo de la selección de exones en la secuencia del ARNm por el mecanismo de corte-empalme, que elimina selectivamente algunos exones (splicing alternativo), que al traducirlos a proteínas dan lugar a isoformas. Muchas proteínas sufren, además, modificaciones postraduccionales (MPT) o forman oligómeros con otras proteínas. Así, dependiendo de sus MPT, un gen puede generar proteínas diferentes, con distintas funciones.
El proteoma es el complemento proteínico total expresado por un genoma, que identifica el grado de expresión, isoformas, MPT, interacciones y localización de las proteínas, con lo que contribuye al conocimiento de las enfermedades complejas. El proteoma es dinámico y varía con las condiciones ambientales de célula, tejido y organismo. La comparación de patrones proteómicos del suero y líquido sinovial de pacientes con AR permite diseñar nuevas herramientas diagnósticas y para seguir la respuesta al tratamiento y, eventualmente, terapias específicas.
Una búsqueda en la base de datos de NCBI con las palabras “arthritis” y “proteomics” devolvió 54 publicaciones hasta agosto de 2007, mientras que una búsqueda más restringida con las palabras “rheumatoid”, “arthritis” y “proteomics” reveló 36 publicaciones. Como comparación, cuando se utilizaron las palabras “cancer” y “proteomics” se obtuvieron 1.990 publicaciones.
Los estudios de proteómica en suero y liquido sinovial de diferentes enfermedades reumáticas identificaron proteínas de fase aguda83. Por ejemplo, en la AR los niveles de PCR indican el grado de inflamación, pero no son indicativos de la severidad de la enfermedad ni son suficientemente sensibles para medir la respuesta al tratamiento84–86. La SAP está presente en suero, plasma y líquido sinovial de pacientes con AR, pero no en osteoartritis (OA)87. Algunas isoformas del fibrinógeno y de las calgranulinas A, B y C se asocian a AR88 y estas últimas también a espondiloartropatías, pero ninguna a OA83.
Liau et al identificaron 33 posibles proteínas marcadoras en líquido sinovial de pacientes con AR89,90, incluidos miembros de la familia de proteínas S100 y otras proteínas, como osteopontina91, ciclofilina (el blanco de la ciclosporina A), catepsina B y otras que se han encontrado elevadas en líquido sinovial de pacientes con AR y erosiones.
La proteómica también permite identificar los patrones de autoanticuerpos, que en individuos sanos podrían predecir una enfermedad autoinmunitaria. Además, los perfiles de autoanticuerpos en AR también podrían predecir el curso de una enfermedad ya diagnosticada92. A manera de resumen, la AR es una enfermedad que afecta principalmente a individuos en etapas productivas de la vida. Su prevalencia del 0,1 al 0,5% predice que en el mundo hay más de 100 millones de individuos afectados por esta devastadora enfermedad, en el 10% sigue un curso agresivo que no responde a tratamiento convencional, con costos enormes en cualquier sentido. Por ello es fundamental optimizar los medios de diagnóstico, pronóstico y evaluación de respuesta terapéutica, lo que implica, sin duda alguna, el conocimiento de sus mecanismos patogénicos.
El conocimiento de las variantes genéticas de la AR, aplicado individualmente, permitirá predecir qué pacientes responderán y quiénes tendrán resistencia a distintos agentes terapéuticos, permitiendo asignarlos al mejor tratamiento posible, con menos fracasos terapéuticos (un 50% menos), tiempo de respuesta corto, mayor posibilidad de remisión y, consecuentemente, menos daño estructural. El uso sistemático de patrones genéticos y/o proteómicos servirá también para el diseño de nuevos tratamientos. Los cambios en el patrón proteómico podrían facilitar el seguimiento del comportamiento biológico de la AR conforme se modifica la actividad.


