



A case of destructive arthropathy of hips and shoulders with tumoral calcinosis associated with calcium oxalate deposits in a patient with primary oxalosis and end stage renal disease on hemodialysis.
Se presenta el caso de artropatía destructiva de caderas y hombros con calcinosis tumoral asociada a depósitos de oxalato de calcio en un paciente con oxalosis primaria e insuficiencia renal terminal en hemodiálisis.
Oxalate crystal deposition disease is a rare condition seen primarily in patients with primary hyperoxaluria (PHO) and oxalate nephropathy and forms of secondary oxalosis in patients with ESRD on chronic dialysis, oxalate supplementation, thiamine deficiency and pyridoxine or oxalate formation due to Aspergillus niger. Musculoskeletal manifestations of calcium oxalate deposition disease are similar to those presented in calcium pyrophosphate crystals arthropathies. We report the case of a patient with arthropathy and tumoral calcinosis and associated deposits of calcium oxalate.
Case DescriptionThe case is a 22-year-old Latin American Mestizo O positive male, with no toxic habits. The patient had no family history of kidney stones. He was diagnosed with short stature at age 6. At age 8 he presented repeated episodes of kidney stones and at 10 was diagnosed with primary hyperoxaluria, with progression to ESRD. At 12 he underwent renal replacement therapy with peritoneal dialysis and at 13 underwent living donor transplantation with graft loss after 5 months, so hemodialysis was started at a rate of 3 sessions a week and has remained so since then. 2 years after starting hemodialysis he presented bone pain and carpal, metacarpophalangeal and bilateral knee symmetrical and additive arthritis. At 16 he had a pathological fracture of the right femur, which required open reduction and internal fixation. The bone pain and polyarthritis followed a progressive course, with no response to treatment with non steroidal anti-inflammatory drugs (NSAID), and the patient noted the development of tumors located in soft tissue, which prevented gait at age 19. The symptoms persisted despite infiltration with glucocorticoids, low-dose oral steroids, opioid analgesics and NSAID. During the patients latest assessment we found that the patient had low height, pectus carinatum, short limbs, wrist and ankle subluxation and periarticular tumors located on shoulders, hips and knees, at the expense of soft tissue.
Laboratory studies showed: Vitamin A 7.1ng/ml (low <10), ferritin 1342ng/ml (30–400), cortisol 7.87mg/dl (5–25) parathyroid hormone (PTH) 4.08pg/ml (10–65), iron saturation percentage of 101% (15–55), iron binding capacity without saturating 63g/dl (250–450), iron 64mg/dl (50–170mg/dl), creatinine 3.4mg/dl (0.4–1.2), BUN 36.45mg/dl (5–23), glucose 83mg/dl, calcium 9.7mg/dl (8.4–10.2), phosphorous 5.1mg/dl (2.7–4.5), magnesium 2.4mg/dl (1.6–2.6), alkaline phosphatase, 296U/l (40–129) LDH 507U/l (240–480), gamma glutamyl transferase 640U/l (10–71), alanine aminotransferase 23U/l (2–41), aspartate transaminase 50U/l (2–38), albumin 2.3g/dl (3.4–4.8) and uric acid 6.5mg/dl (2.4–7).
An abdominal ultrasound showed both kidneys to be hypoplastic with increased echogenicity and renal calcifications, with hepatic and spleen enlargement and a normal pancreas. An abdominopelvic computed tomography showed nephrocalcinosis, hepatomegaly, splenomegaly, arteriosclerosis and, osteosclerosis of vertebral and pelvic bones.
A transthoracic echocardiogram showed a systolic pressure of 61mmHg in the pulmonary artery, normal left ventricular diameter, a thickened wall, normal mobility, mild dilated right chambers, valvular sclerosis, mild mitral regurgitation and moderate tricuspid regurgitation.
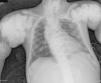
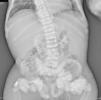
The chest X-ray (Fig. 1) demonstrated nodular periarticular calcification in the shoulders, dorsal vertebral osteosclerosis and vertebral collapse of thoracic vertebrae 5 and 6, with sequelae of rib fractures and a fracture of the right humerus. An abdominal X-ray (Fig. 2) showed bilateral nephrocalcinosis, vertebral osteosclerosis which also affected pelvic bones, bilateral nodular calcifications and a subtrochanteric fracture.
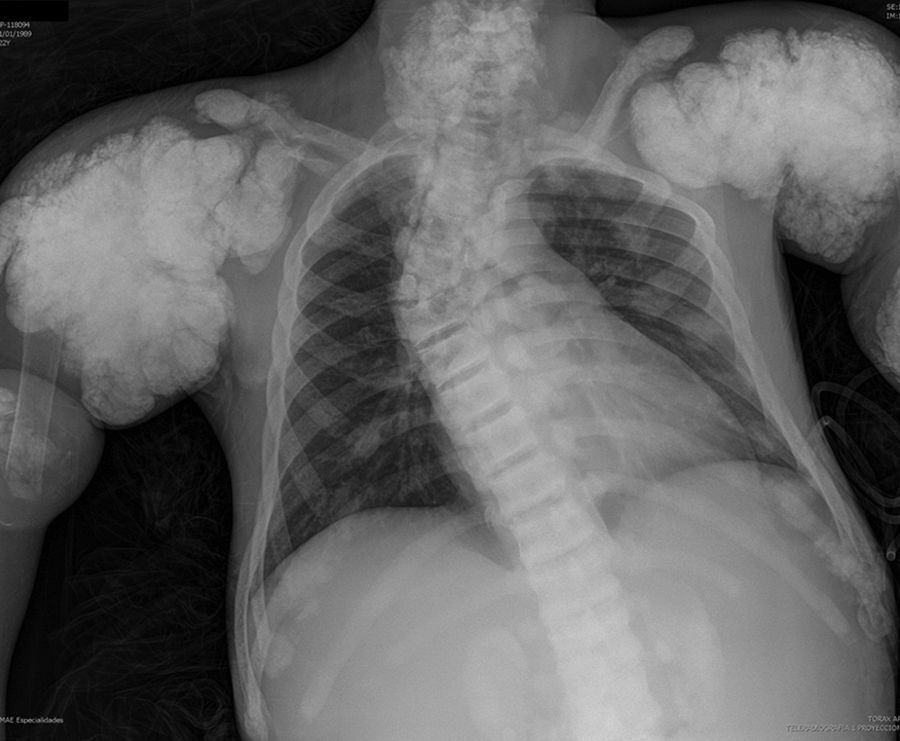
Nodular calcifications of the shoulders. Superior and inferior vertebral plate osteosclerosis (rugby jersey spine) and vertebral collapse of dorsal vertebrae 5 and 6. Bulbous growth on the ends of the ribs and clavicles. Osteosclerosis of clavicles and rib fracture sequelae. Fracture of the right humerus.
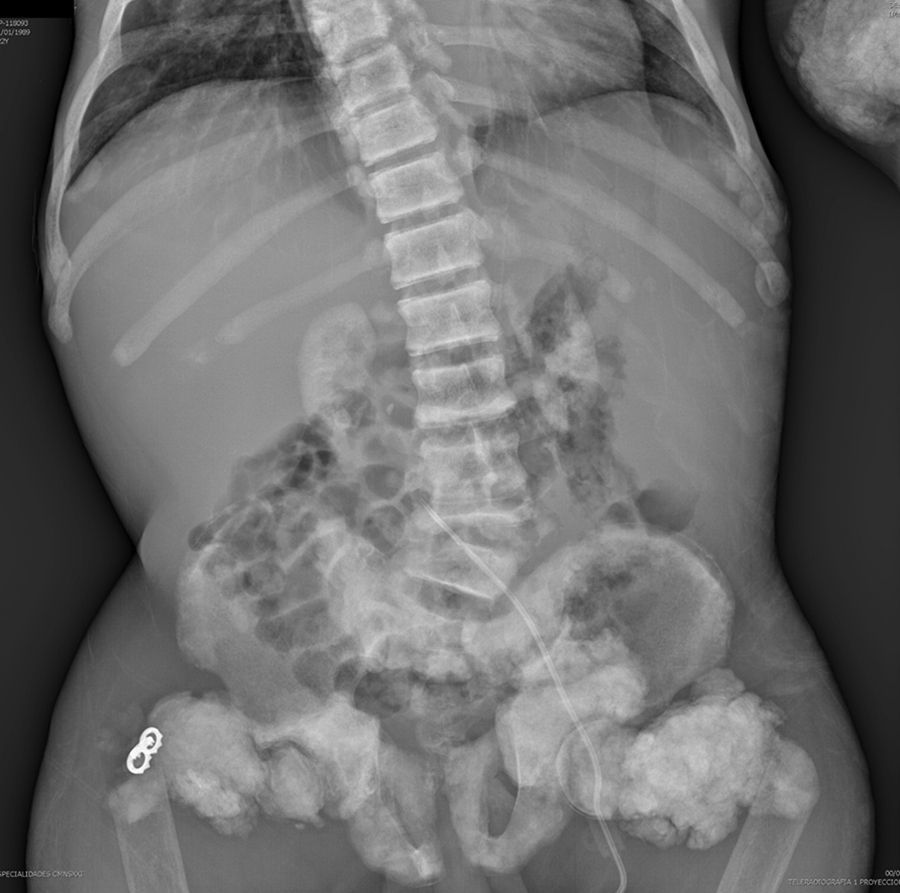
PHO is an autosomal recessive inborn error of metabolism leading to an enzyme deficiency of alanine-glyoxylate aminotransferase in hepatic peroxisomes. The enzyme deficiency causes an overproduction of oxalate which is eliminated by the kidneys and precipitates forming crystals that are deposited in various tissues. PHO diagnosis is performed before the age of 5 in 65% of cases. The main cause of death is uremia, which in 80% of cases occurs before age 20.1
Since oxalate is eliminated through the kidney, this is the first and primary target organ, leading to the appearance of repeated stone formation in the first decades of life, nephrocalcinosis and early renal failure.2 When terminal renal failure occurs and oxalic acid cannot be excreted, rapidly evolving tissue deposits develop particularly in the kidneys and skeleton.2,3 Bones are one of the main affected organs, with unusually serious lesions having been described, especially in patients with chronic renal failure on dialysis.4 Oxalate deposits and the surrounding granulomatous reaction induce lesions similar to secondary hiperparatiroidism which are particularly serious.4
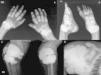
The pattern of joint involvement is more commonly acute or chronic symmetrical polyarthritis or oligoarthritis, with involvement of the metacarpophalangeal and proximal interphalangeal joints, with or without tenosynovitis, along with miliary or cottony skin and finger arterial calcifications5,6 (Fig. 3), but can occur in other joints, such as knees, elbows, ankles and the first metatarsophalangeal joint. In autopsy studies, calcium oxalate deposits in joint tissue and bone oxalosis occur in approximately 90% of patients with renal insufficiency undergoing chronic hemodialysis.6
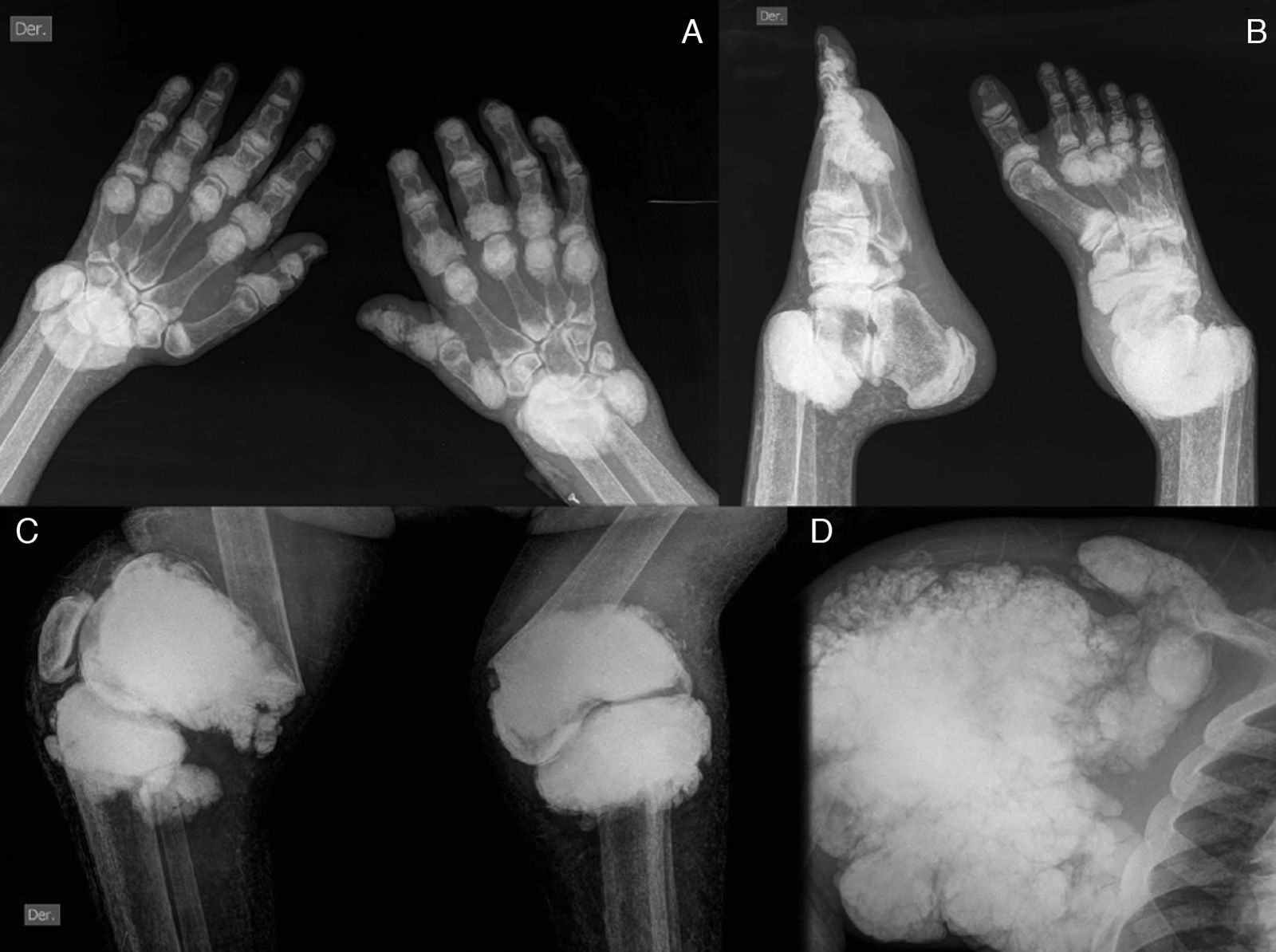
(A) X-ray of hands with periarticular calcinosis on interphalangeal, metacarpophalangeal and wrist joints, decreased bone density, resorption of the distal epiphysis of the radius and ulna. (B) X-ray showing periarticular calcinosis on interphalangeal and metatarsophalangeal tibiotalar joints, osteosclerosis of the phalanges and metatarsal tibiotalar joints dislocation and bone resorption of the distal epiphysis of the tibia and fibula. (C) X-ray of the knees with periarticular tumoral calcinosis, bilateral supracondylar fracture with medial deviation of the femoral shaft. (D) X-ray of shoulder with periarticular tumoral calcinosis, resorption of the humeral head and neck, clavicle bone resorption.
El Hage et al. conducted a review of 12 consecutive patients with type 1 PHO, all with renal involvement, 4 with ESRD undergoing dyalisis.7 The main symptom was bone pain and was present in only 4 of the severely involved patients and appeared in the second year of dialysis. The 2 most severely affected patients had evidence of pathologic fractures.7
Kidney damage is usually the result of a combination of nephrolithiasis, secondary nephrocalcinosis and interstitial fibrosis. Renal failure is associated with the rapid deposit of the crystals in the kidney, myocardium, skin, blood vessels and bones; when the glomerular filtration rate decreases below 30–40ml/min/1.73m2, oxalate cannot be efficiently excreted by the kidneys and reaches saturation levels.8 Oxalate saturation depends on serum levels and these are inversely related to the glomerular filtration rate.9 This saturation, which occurs almost universally in the serum of patients with terminal uremic PHO, causes the systemic oxalosis affecting patients on dialysis.2 In the work of Worcester et al. the saturation threshold for serum oxalate was found to be 40–50μUmol/l, and a threshold was reached with serum creatinine levels of about 9mg/dl2. Other studies and follow up of patients with renal oxalosis and renal failure have reported that patients with predialysis renal failure have no clinical or radiological manifestations of bone disease.
Typical locations of crystal deposits in the skeleton are the segments of the metaphysis of tubular bones.10 The distribution of crystals in bone at sites of endochondral or intramembranous ossification suggests precipitation in vascularized areas when levels are high. Bone oxalosis may be due to the combination of hyperparathyroidism, renal osteodystrophy and the inflammatory response induced by calcium oxalate crystals.11,12
As regular dialysis treatment can prolong survival of patients, a new syndrome, characterized by intense and continuous deposition of calcium oxalate crystals in the soft tissues and bones can develop.13 The appearance of bone problems in dialysis patients demonstrates that dialysis is less effective in removing oxalate than the healthy kidney. Likewise, cases of oxalosis secondary to dialysis have been reported and chronic renal failure may lead to serum oxalate levels 4–8 times higher than normal, resulting in inefficient removal of oxalate by hemodialysis and peritoneal dialysis.8 The type of dialysis also affects the rate of deposition of crystals: an oxalic acid clearance of 80ml/min by hemodialysis has been reported, but is only 6ml/min in the case of peritoneal dialysis.14 Marangella et al. studied oxalate balance in PHO patients undergoing hemodialysis and the daily generation rate in the study was 360–630mg and removal during dialysis did not reach 30% of the amount generated, resulting in a daily calcium oxalate deposits of 180–360mg; to achieve balance in dialysis oxalate, sessions should last from 13 to 15h, resulting impracticable.2,15
Imaging examination may reveal small kidneys, with parenchymal calcifications. The spectrum of radiologic abnormalities in the skeleton is the result of changes caused by the deposition of oxalate in bone and renal osteodystrophy in patients with chronic renal insufficiency.3
Radiological signs can be divided into those with greater specificity and those with less specificity. More specific imaging signs of oxalosis are found mainly in severely affected patients and include irregular sclerotic transverse bands in the metaphyseal segments of tubular bones (femur, humerus, tibia, fibula, metacarpals, metatarsals and phalanges), radiolucent metaphyseal bands and osteosclerosis of vertebral bodies which involve first the upper and then the lower vertebral endplates, creating the appearance of a rugby jersey before spreading to the rest of the vertebral body and the appearance of the “bone in bone” typical image.7 Metaphyseal bands and sclerotic areas of the vertebral bodies are not specific for oxalosis and differential diagnosis includes, among others, lead poisoning, leukemia and thalassemia.1 Other, less specific findings include clubbing of the metacarpal bones, sclerosis of the clavicles, a radiolucent or radiodense rim around the epiphyses, carpal and tarsal bones, cystic bone changes, epiphyseal invagination, bulbous growth of the ends of the ribs and clavicles, osteosclerosis in patches (Paget-like), subperiosteal resorption and pathological fractures. Day et al. found that bone radiographic abnormalities depend on the patient's age at the time of occurrence of renal failure and the degree of renal transplant success; the most characteristic skeletal changes occurred in 6 of 7 patients who developed renal disease before age 7.
Soft tissue calcifications are a frequent complication in patients with chronic renal failure. The term tumoral calcinosis refers to massive deposit of calcium salts that form multiloculated tumors around joints. The pathogenesis of the skeletal and soft tissue alterations associated with chronic renal failure is multifactorial and, although not entirely clear, there are two main factors that work together in its development: secondary hyperparathyroidism and an altered metabolism of vitamin D.
Soft tissue calcification may be seen in up to 79% of patients on dialysis. Chronic renal insufficiency leads to secondary hyperparathyroidism which causes release of calcium from bones and inhibition of the tubular phosphate reabsorption area. When the product of calcium×phosphorus is high, in the order of 65–70 (usually 40), subcutaneous deposition of calcium phosphate occurs.16
Idiopathic tumoral calcinosis is characterized by the development of large amorphous calcium phosphate deposits around large joints, elevated levels of normal serum phosphorus and PTH levels. The most likely cause is an increase in the tubular reabsorption of phosphate.17 These lesions are usually asymptomatic and may rarely produce symptoms of compression of adjacent structures.
Tumoral calcinosis associated with uremia is a rare complication seen in patients on long term dialysis and has a multifactorial etiology; it is characterized by deposits of calcium salts in periarticular areas, sometimes in mass. The most important etiopathogenic factor is the deterioration of the product of serum calcium x phosphorus; hyperparathyroidism and periarticular tumors frequently coexist without joint involvement or invasive process.18,19 Clinically, patients present periarticular painless firm masses which show progressive growth. This growth is expansive, and there is no visceral, muscle or bone invasion. Predisposing factors are severe hyperparathyroidism and a calcium×phosphorus product greater than 70.18 In the absence of secondary hyperparathyroidism, the elevation of the calcium–phosphorus product is originated by iatrogenic hypercalcemia and/or multifactorial hyperphosphatemia: excessive and prolonged administration of calcium carbonate and calcitriol, inadequate intake of phosphate binders and insufficient dialysis.18
Chronic ingestion of high doses of vitamin D can cause hypervitaminosis D; its symptoms and signs are due to hypercalcemia and include weakness, headache, nausea, polyuria and nephrolithiasis.16
In our case we ruled out the above-described factors as inducers of calcifications. Vitamin D levels were below the reference range, which rules out hypervitaminosis D. We did not document secondary hyperparathyroidism; by contrast, PTH levels were found suppressed, and we did not find elevated levels of calcium–phosphorous (about 50) to justify tumoral calcinosis. In addition, the most common skeletal manifestation of hypoparathyroidism is osteosclerosis, with radio opaque bands in the metaphyses of long bones and, although subcutaneous calcifications may be seen especially in the hips and shoulders, these are asymptomatic; likewise the patient did not present hypocalcemia or neuromuscular signs to support the diagnosis of hypoparathyroidism. For technical reasons at baseline and due to the subsequent loss of patient follow-up, it was not possible to perform bone biopsies and the study of crystals in synovial fluid, which would have helped clarify the pathogenesis of the disease.
Our patient was clinically compatible with primary hyperoxaluria and presenting recurrent nephrolithiasis, nephrocalcinosis with childhood onset progressing to ESRD; the key was the diagnosis of hyperoxaluria, but this data are only useful in the absence of renal failure, which was not determined. The absence of musculoskeletal symptoms before dialysis is consistent with reports that patients with PHO in a predialysis stage are asymptomatic, without radiographic bone oxalosis, and in a period of 1–2 years after dialysis, most evolved with progressive bone pain, which can produce severe bone disease with skeletal deformities, spontaneous fractures and disability.4 The longer survival of our patient due to renal replacement therapy, with increased exposure time and precipitation of oxalate crystals at the osteoarticular level, is considered the main factor for late changes in bone oxalosis, the presence of pathological fractures and tumoral calcinosis. In a series of more than 200 patients on dialysis and serum PTH levels above those of our patient, vertebral or pelvic osteosclerosis is not usually found, whereby this pattern is readily attributable solely to renal osteodystrophy.13 Lab data found moderately elevated alkaline phosphatase, which has been described in the course of patients with oxalosis mainly in those undergoing hemodialysis.4
Brancaccio et al. reported that the deposition of oxalate and hyperparathyroidism is involved in the genesis of bone lesions, but the former is much more important, in patients with PHO and hyperparathyroidism who undergo parathyroidectomy, bone lesions progress despite normal serum PTH levels and threshold serum calcium levels, indicative of bone resorption independent of PTH.13
As a result, oxalosis secondary to hemodialysis combined with primary hyperoxaluria could have aggravated the skeletal changes in our patient. Furthermore, most of the changes in the patient's bones could be the result of the combined effects of oxalate crystal deposition and progressive development of renal osteodystrophy. However, skeletal changes are more likely to be related to oxalosis. It seems logical to consider oxalate deposits as the main cause of severe bone lesions and the tumoral calcinosis observed in this patient.
The aim of this article was to describe the radiographic spectrum of bone oxalosis in the major joints. To our knowledge, no other article mentions a destructive arthropathy disease associated with this complex, which can be due to the increased survival of patients on hemodialysis. A comprehensive literature search was unable to retrieve any items that describe a relationship between oxalosis and destructive arthropathy of the shoulders, hips and knees.
ConclusionThe spectrum of radiographic bone changes in primary hyperoxaluria and oxalosis are due to many factors that influence bone metabolism in this complex disease, including high levels of calcium oxalate, secondary hyperparathyroidism and renal osteodystrophy. We described the case of a patient with destructive arthropathy of the large joints, in whom we reasonably ruled out hyperparathyroidism, hypoparathyroidism and related phosphocalcic disorders, suggesting deposition of oxalate as the cause of his arthropathy.
Ethical ResponsibilitiesProtection of People and AnimalsThe authors state that no experiments were performed on humans or animals.
Data ConfidentialityThe authors state that no patient data appear in this article.
Right to Privacy and Informed ConsentThe authors have obtained informed consent from patients and/or subjects referred to in the article. This document is in the possession of the author of correspondence.
Conflict of InterestThe authors declare no conflict of interest.
Please cite this article as: Horta-Baas G, et al. Artropatía destructiva de grandes articulaciones y calcinosis tumoral asociada a oxalosis primaria: reporte de un caso y revisión de la literatura. Reumatol Clin. 2013;9:181–5.


