



La artritis reumatoide ha experimentado en la última década una revolución terapéutica, derivada del conocimiento de los procesos patogénicos y favorecida por el desarrollo de la tecnología necesaria para distribuir tratamientos moleculares. Las nuevas terapias permiten diferenciar subtipos de pacientes según la respuesta clínica y además mejoran nuestra comprensión de la enfermedad. Ello hace vaticinar la llegada de nuevas generaciones de moléculas para un tratamiento individualizado. Uno de los campos hacia donde se dirigen las investigaciones es la epigenética. Los mecanismos de regulación epigenéticos son interruptores que deciden cuándo y cómo expresar determinados genes en cada célula. Actuando como vigilantes de una expresión génica inapropiada, protegen al organismo del desarrollo de tumores. La principal ventaja de los tratamientos epigenéticos podría ser su selectividad por las células que muestran patrones epigenéticos alterados, por lo que el reto es identificar estas alteraciones entre los pacientes con artritis reumatoide. Aunque debe establecerse su perfil de seguridad, parece probable el uso de terapias epigenéticas en las enfermedades autoinmunes.
Over the last decade, the management of rheumatoid arthritis has evolved as a result of both the understanding of disease-related processes and the availability of the necessary high-throughput technology to provide patients with molecule-based therapies. New therapies allow the classification of patients into subsets as regards clinical response, at the same time adding to our knowledge of rheumatoid arthritis pathogenesis. New generations of molecules will likely soon be ready for “a la carte” treatment of patients. A promising field of research is epigenetics. Epigenetic regulatory mechanisms switch on and off the transcription of specific genes in individual cells. Acting as observers on non-adequate gene expression, these mechanisms yield protection against the development of tumours. The major achievement of epigenetic therapies could be their selective action on cells with altered epigenetic programs, and it is our challenge to recognize these alterations among patients with rheumatoid arthritis. Although safety concerns may arise, epigenetic drugs will likely be used to treat autoimmune diseases.
Los avances recientes en el conocimiento de los mecanismos de regulación de la expresión génica han cambiado nuestra concepción de las enfermedades complejas, como el cáncer, el lupus o la artritis reumatoide (AR). Estos rasgos poligénicos se asocian a un conjunto de genes de baja penetrancia, los cuales confieren susceptibilidad o protección y se transmiten con un patrón de herencia no Mendeliano1 En el caso de la AR, se desconocen en gran medida los factores ambientales que puedan participar en su aparición, lo que incrementa la dificultad para predecir su desarrollo. Lo más probable es que los pacientes que buscan atención médica sean tan solo una pequeña parte de la población artrítica. Sin embargo, sería de gran interés estudiar también a los pacientes con enfermedad leve de curso remitente, para poder identificar los factores que determinan la progresión. En este sentido, las enfermedades complejas se caracterizan por un amplio espectro de gravedad, que refleja el efecto acumulativo de diferentes factores permisivos.
Independientemente de la carga genética, la actividad transcripcional de los genes está exquisitamente regulada de forma específica en cada tejido, en función del estado evolutivo. Durante el desarrollo embrionario se lleva a cabo una represión en la expresión de gran cantidad de genes, que va progresando a lo largo de las divisiones celulares y es responsable de la especificación2. Esto se ejecuta mediante un programa de silenciamiento epigenético, un término acuñado para subrayar que se produce una supresión estable de la expresión génica, generando patrones transmisibles a las células hijas, pero sin que ello conlleve cambios en la línea germinal3. Aunque por el momento, el alcance de las modificaciones epigenéticas no se conoce en toda su extensión, es ya evidente que procesos fundamentales como el crecimiento celular, la secreción hormonal, la respuesta inflamatoria y la maduración de las células inmunes están gobernados por este tipo de regulación3. Del mismo modo, las enfermedades degenerativas, la inflamación o el cáncer, se caracterizan por la acumulación de marcas epigenéticas en las células enfermas. Ello ha llevado a postular que distintos factores ambientales pasados y recientes conducen a la alteración de los patrones de expresión génica por mecanismos epigenéticos, pudiendo éstos, en consecuencia, explicar en gran medida la contribución ambiental al desarrollo de las enfermedades complejas4.
Mecanismos epigenéticos de silenciamiento transcripcionalLa inhibición de la transcripción génica se produce mediante la metilación del ADN y los cambios en la conformación de la cromatina. Ambos mecanismos actúan de forma coordinada y afectan a la accesibilidad de los genes para su lectura (fig. 1)4,5. La cromatina está estabilizada por las uniones covalentes entre el ADN y las histonas y otras proteínas básicas del núcleo celular. Las modificaciones postraducción de las histonas dan lugar a cambios de afinidad en su unión con el ADN, generando una conformación cerrada o abierta de la cromatina (hetero o eucromatina, respectivamente). Tanto la metilación del ADN como las modificaciones de las histonas son procesos reversibles, catalizados por enzimas y cofactores específicos. Ello indica que las células son capaces de cambiar sus patrones de expresión epigenéticos en respuesta a diferentes estímulos, permitiendo al mismo tiempo su manipulación con intención terapéutica. Sin embargo, muchas de estas marcas epigenéticas tendrán que esperar a la siguiente división mitótica para poder ser modificadas.
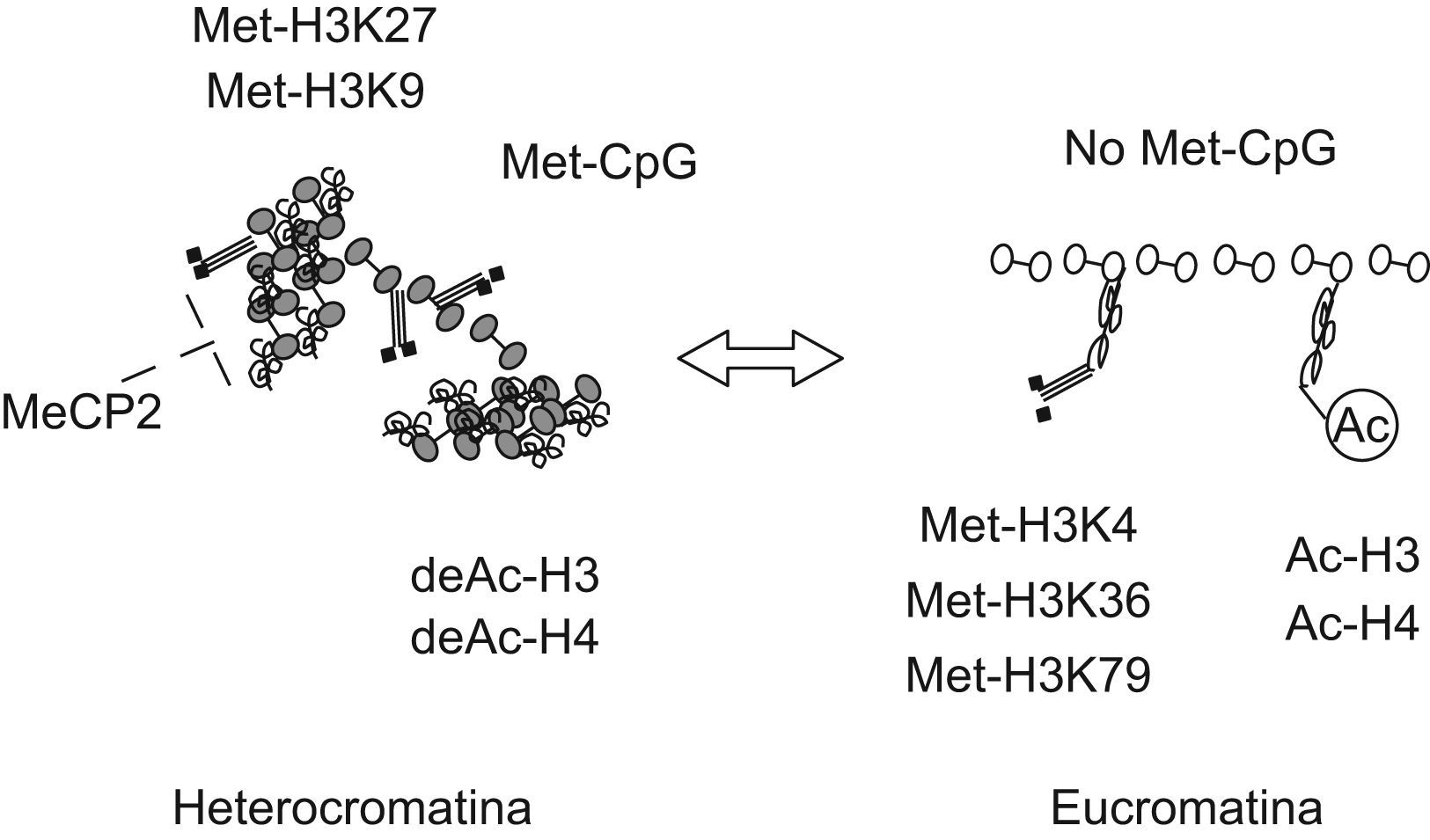
Marcas epigenéticas asociadas a las conformaciones cerrada y abierta de la cromatina. El silenciamiento génico se consigue con un alto nivel de metilación del ADN (Met-Dinucleotidos citosinafosfoguanina), en combinación con la deacetilación de las histonas H3 y H4 (deAc-H3, deAc-H4) y la metilación de la histona H3 en los residuos lisina 9 y 27 (Met-H3K9, Met-H3K27). Mediante estos cambios en su estructura, las interacciones entre las histonas y el ADN son más fuertes, determinando una conformación cerrada o heterocromatina. La presencia de moléculas capaces de estabilizar las uniones covalentes entre el ADN metilado y las histonas, como MeCP2, producen un empaquetamiento aún más denso del genoma. La activación de la expresión génica se caracteriza por la acetilación de las histonas H3 y H4 (Ac-H3, Ac-H4) y la metilación de las lisinas en posición 4, 36 y 79 de la histona H3 (Met-H3K4, Met-H3K36, Met-H3K79). Ello determina una configuración abierta, con uniones laxas con el ADN, clásicamente denominada eucromatina. Los cambios entre ambas configuraciones son, en gran medida, dinámicos.
La metilación del ADN tiene lugar en determinadas regiones ricas en dinucleotidos citosinafosfoguanina por la acción de las ADN metiltransferasas (DNMT). Las histonas H3 y H4, por su parte, pueden sufrir numerosas transformaciones, como fosforilación, metilación, acetilación, ribosilación, sumoilación, ubiquitinación, así como las reacciones inversas y varias combinaciones. La mayoría de ellas se producen en los aminoácidos lisina (K) o arginina (D) cercanos al extremo amino-terminal de la proteína. Existe una larga lista de enzimas que participan en las modificaciones de las histonas, en la que destacan las familias de las histona-acetilasas (HAT) y las histona-deacetilasas (HDAC)6 La eliminación de grupos acetilo se relaciona estrechamente con el silencio transcripcional, mientras que la acetilación abre la conformación de la cromatina y se asocia a un incremento de la expresión génica (fig. 1)7,8.
Los mecanismos epigenéticos de la artritis reumatoideLas respuestas inflamatorias en la septicemia y en la AR en brote muestran una explosión inicial de citoquinas de características muy similares; sin embargo, en la septicemia, esta oleada deja paso a un descenso en los niveles de las citoquinas que no tiene lugar en la artritis9 Esto indica que existe un control defectuoso en la finalización del proceso en los pacientes con artritis. La principal ruta encaminada a la producción de las citoquinas proinflamatorias, tanto durante la sepsis como en la AR, es la vía de activación innata del factor de transcripción (NF)κB10. Esta ruta es un claro ejemplo de cómo los mecanismos epigenéticos intervienen en la regulación de las respuestas celulares. El NFκB se encuentra en estado latente en el citoplasma de las células, anclado a su inhibidor, IκB. A su vez, la disponibilidad del inhibidor está regulada mediante la metilación de su promotor, situación que puede determinar importantes diferencias en la producción de citoquinas. Por otra parte, la cromatina está plegada en una conformación cerrada en las regiones correspondientes a los promotores de las citoquinas pro-inflamatorias. En sincronización con la translocación al núcleo del NFκB, han de producirse cambios transitorios en esta conformación (una demetilación en K9 y una fosforilación en la serina (S)10 de la histona H3)11,12 para permitir la incorporación de la maquinaria transcripcional a sus sitios de unión en los promotores del factor de necrosis tumoral (TNF)α y la interleuquina (IL)1β13
Otro de los procesos estrechamente regulado por mecanismos epigenéticos es la diferenciación de las células inmunocompetentes14 La AR se caracteriza por la polarización hacia respuestas Th1 y Th17. La diferenciación de las células T doble-positivas hacia las Th1 se produce a través de la metilación progresiva del promotor de la IL4, que es la citoquina específica de las células Th2, hasta su supresión completa15. Por su parte, el factor de transcripción FoxP3, definitorio de las funciones T reguladoras, es susceptible de silenciamiento por metilación, un evento que inclina la balanza hacia la producción de células Th17 y favorece la autoinmunidad16. Estos cambios en el fenotipo de las células T son, en gran medida, reversibles y existe evidencia de que algunas citoquinas, como el factor de crecimiento transformante (TGF)β, la IL6 y el interferón (IFN)γ, participan de forma activa en el establecimiento de los patrones epigenéticos en las subpoblaciones de células T17.
El fenotipo invasivoLa primera información acerca de la implicación epigenética en patología humana provino de los tumores, donde se objetivó que la presencia de determinadas marcas epigenéticas promovía la invasividad y la supervivencia de las células afectadas. En particular, un evento típico de las fases iniciales en la tumorigénesis es la metilación de novo de genes reguladores o supresores tumorales18 En este sentido, aunque las DNMT son esenciales para adquirir los patrones normales de metilación génica, sus inhibidores se han revelado como una estrategia interesante para hacer a las células reexpresar los genes anormalmente hiper-metilados19 Los primeros agentes demetilantes empleados en el tratamiento de enfermedades humanas fueron la 5-aza citidina y la 5-aza-2′-desoxicitidina (5-aza CR y 5-aza CdR), seguidos por los menos tóxicos Zebularina y MG98. También se ha constatado el papel de las HDAC en el silenciamiento génico asociado a la des-diferenciación de algunos tumores y a su resistencia a la quimioterapia. La suberoil anilida del ácido hidroxámico (SAHA) es un inhibidor natural de la actividad de las HDAC que está aprobado para el tratamiento de la leucemia cutánea de células T, mientras que otros 2 antagonistas de estos enzimas, el ácido fenilbutírico y el ácido valproico, han demostrado su capacidad de corregir los patrones alterados de expresión génica en diferentes enfermedades20.
El grado de metilación del genoma en la membrana sinovial reumatoide está siendo objeto de estudio, dada la similitud entre los fibroblastos locales y las células transformadas. Aunque desprovistos de mutaciones, los fibroblastos sinoviales (FS) de los pacientes muestran una supervivencia anormalmente elevada y ejecutan un programa invasivo, caracterizado por la secreción constitutiva de proteasas y citoquinas21,22. En las articulaciones de los pacientes, las células que se hallan en áreas de invasión del hueso y el cartílago articular expresan protooncogenes y factores de crecimiento23,24. Este patrón locorregional en la expresión génica sugiere que desde el entorno inflamatorio se podría desencadenar un determinado epigenoma que se propaga a la progenie de las células modificadas. Parte de estos mecanismos se está empezando a desentrañar. Por ejemplo, la longevidad que caracteriza a los FS en la AR se ha puesto en relación con un alto grado de metilación en el promotor de moléculas proapoptóticas, como el receptor letal DR3,25. Del mismo modo, se ha demostrado que la Tricostatina A (TSA), un inhibidor de las HDAC, induce la reexpresión de otras moléculas proapoptóticas en estas células, incrementando consecuentemente su senescencia26,27. Estos hallazgos sugieren que las células sinoviales en la AR muestran un programa de silenciamiento génico alterado, que podría estar en relación con su tendencia destructiva y que es susceptible de reprogramación.
Pros y contras de las terapias epigenéticas y su posible uso en la artritis reumatoideAún en espera de comprobar el perfil de seguridad que muestran los tratamientos epigenéticos en los pacientes con cáncer, podemos pronosticar que estos tratamientos se introducirán en enfermedades no neoplásicas, como la AR, en un futuro próximo28. La meta principal de los abordajes epigenéticos en la AR será aumentar la expresión de genes reguladores que se encuentren anormalmente silenciados, con la pretensión de atenuar con ello la expresión clínica de la enfermedad. Esto puede conseguirse bien inhibiendo la metilación del ADN o cambiando la conformación de la cromatina en lugares determinados. Como principal contrapartida, el uso de estas estrategias conlleva el riesgo de eliminar marcas epigenéticas constitutivas, como las que evitan la expresión de transgenes, en las células con alto índice replicativo19.
Agentes demetilantesEstos son, con diferencia, los fármacos epigenéticos más prometedores para el tratamiento de la AR. Es probable que la metilación de novo de ciertos genes reguladores favorezca la progresión de la AR y su reexpresión dé lugar a un fenotipo más leve o mejore la respuesta a fármacos modificadores de evolución (fig. 2A). En realidad, algunos de los fármacos empleados actualmente en el tratamiento de la artritis podrían actuar como agentes demetilantes. Cuando comenzaron a estudiarse las modificaciones epigenéticas en el cáncer, se observó que la mayoría de los agentes antineoplásicos ejercían efectos epigenéticos al emplearse a dosis no citotóxicas4,29. El Metotrexato, como antagonista de la dihidrofolato reductasa, limita la acción de las DNMT (fig. 2B). El ácido fólico actúa como aceptor de grupos metionina de la homocisteina, en presencia de vitamina B12. A su vez, la metionina es el donante de los grupos metilo que las DNMT utilizan para llevar a cabo la metilación de los residuos citosina del ADN30. Esto sugiere que el Metotrexato impide las reacciones de metilación de novo (fig. 2B), una acción que podría explicar parte de su beneficio en la AR. Por otra parte, dada la participación de la IL6 en los procesos de metilación del genoma y en las interacciones con las HDAC31 es probable que su inhibición terapéutica con el uso del Tocilizumab redunde en cambios en los patrones epigenéticos asociados a la enfermedad y en el futuro se puedan asociar estos cambios a grupos de respuesta clínica. Existe la preocupación de que el uso de agentes demetilantes pueda conducir a un aumento en los niveles de las metaloproteinasas 3 y 13, moléculas implicadas en la destrucción articular. Estas moléculas tienen dinucleotidos citosinafosfoguanina en su promotor, susceptibles de metilación. En FS en cultivo, la producción de la metaloproteinasas 3 aumenta tras la exposición a 5-aza CdR, mientras que la expresión incrementada de ambas proteasas en el cartílago artrósico se ha puesto en relación con el bajo grado de metilación de su promotor en los condrocitos enfermos respecto a los sanos32,33. No obstante, todo parece indicar que el umbral necesario para eliminar las marcas epigenéticas constitutivas es mayor que el que se precisa para impedir la metilación de novo.
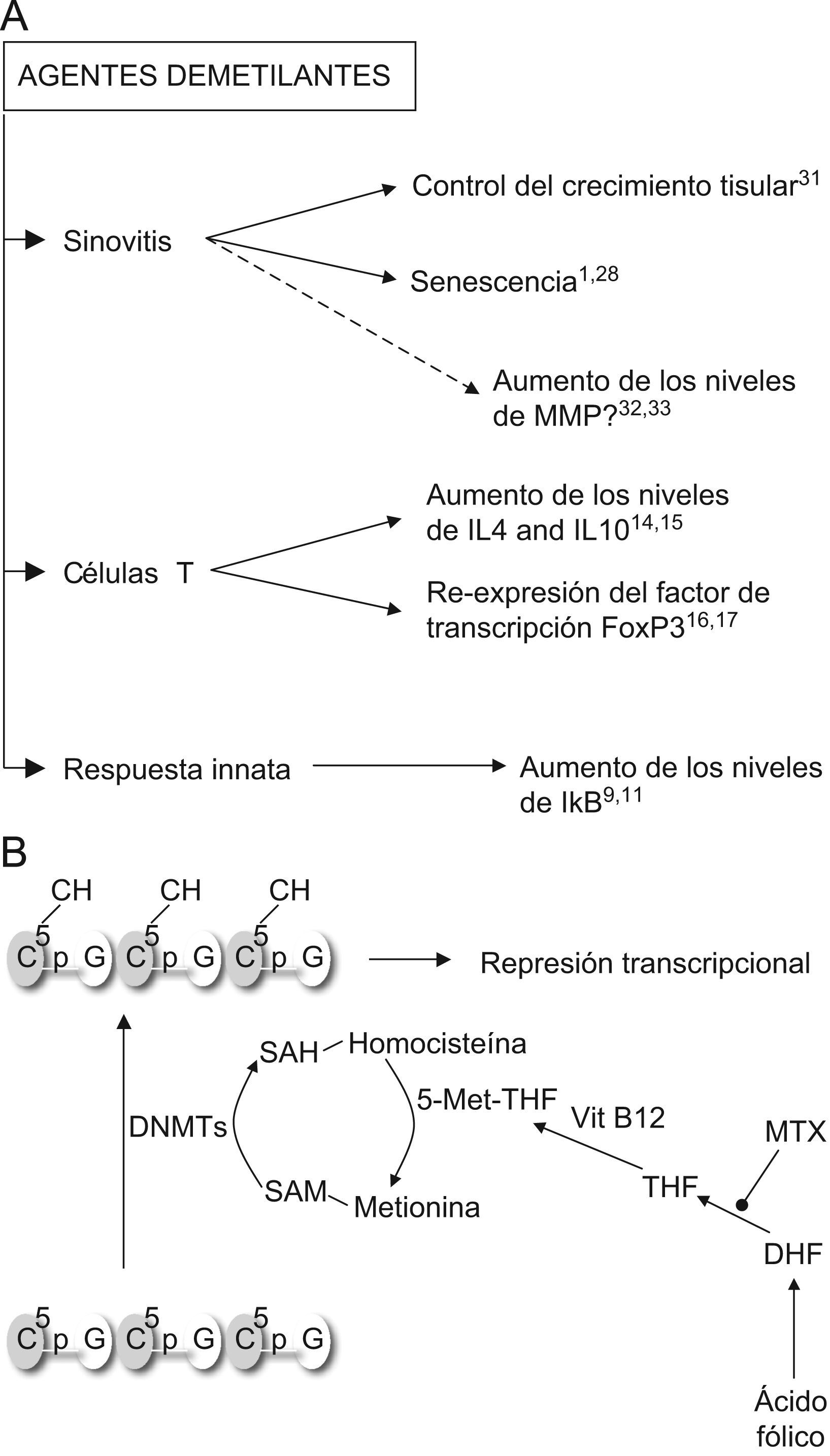
Acciones de los inhibidores de las ADN metiltransferasas. A) Predicción de los efectos del tratamiento con agentes demetilantes en la artritis reumatoide. Según indican los estudios disponibles, el uso de estos tratamientos redundaría en beneficios en los procesos patogénicos principales de la artritis reumatoide: la sinovitis, la polarización de las células T y la inmunidad innata. B) Efecto del Metotrexato (MTX) sobre la acción de las ADN metiltransferasas (DNMTs). DHF: ácido dihidrofólico; THF: ácido tetrahidrofólico; SAM: sulfoadenosil metionina; SAH: sulfoadenosil homocisteina.
Aunque existe una alta expectación con respecto al uso de los inhibidores de las HDAC en el tratamiento de la AR, estos compuestos todavía necesitan demostrar que son candidatos válidos. El FK228 ha demostrado eficacia en varios modelos experimentales similares a la AR, donde fue capaz de reducir la inflamación articular y la tendencia destructiva34. Este beneficio se ha atribuido a un efecto antiangiogénico y a la inducción de moléculas proapoptóticas en el tejido sinovial35. También se han observado efectos beneficiosos con los inhibidores de las HDAC, SAHA y MS-275, en ratones con artritis inducida por colágeno36,37. Sin embargo, es difícil traducir estos efectos a la enfermedad humana, ya que diferentes estudios han sido incapaces de demostrar una actividad elevada de las HDAC en los tejidos sinoviales reumatoides. Por el contrario, parece existir un ratio HAT/HDAC aumentado, relacionado a su vez con la producción de moléculas proinflamatorias, lo que nos hace contemplar el abordaje de la enfermedad con inhibidores de las HDAC con cautela38.
Metiltransferasas de las histonasEl potencial terapéutico de las metiltransferasas de las histonas está aún poco explorado, si bien la participación de las lisina demetilasas LSD1 y Jumonji en la progresión de distintos tumores está bien establecida39. Se trata éste de un campo muy amplio, en el que cada enzima muestra bastante selectividad por modificar residuos concretos40. La familia de lisina demetilasas Jumonji aumenta su expresión en situaciones de hipoxia, por lo que se les supone involucradas en las respuestas inflamatorias derivadas de estrés celular41. En particular, el estado de metilación de los residuos H3K4, H3K9 y H3K27 regula la actividad transcripcional del NFκB, lo que puede ser clave en el diseño de tratamientos42.
Terapias combinadasDado que los sucesos en el núcleo celular tienen lugar en coordinación, y que las modificaciones postraducción no solo afectan a las histonas sino también a los cofactores y los activadores de la transcripción, parece razonable emplear tratamientos epigenéticos en cócteles43. Esta estrategia ha demostrado un sinergismo beneficioso para el abordaje de los tumores, mejorando además el perfil de seguridad, dado que permiten una reducción en las dosis terapéuticas20. El ácido valproico es un ácido graso de cadena corta que actúa como inhibidor de diversas HDAC. Por su parte, el ácido retinoico es capaz de secuestrar y liberar factores de transcripción en el núcleo celular, desplazando a los complejos integrados por las HDAC. La combinación de ambas drogas con la 5-aza-CdR logró la reexpresión de genes reguladores silenciados en células tumorales44 y ha sido ya empleada en pacientes con leucemia aguda mieloide y síndrome mielodisplásico de alto riesgo45. Otro ejemplo de sinergismo es la combinación del inhibidor no selectivo de las HDAC, TSA, con la 5-aza CdR, que dio lugar a la reexpresión de receptores estrogénicos en una línea de cáncer de mama, lo que las sensibilizó a la acción del Tamoxifeno46. Es evidente que se requiere un alto nivel de seguridad para exportar estos tratamientos a patologías no neoplásicas, como la AR, pero parece probable que se puedan preparar combinaciones parecidas a las mencionadas, tras seleccionar los procesos alterados de forma individualizada en los pacientes con AR.
Epílogo: los retos del tratamiento de la artritis reumatoideLa principal lección que nos proporcionan los denominados tratamientos biológicos es que la AR es una entidad heterogénea que requiere un abordaje terapéutico individualizado47 El reto para aplicar el mejor tratamiento a cada caso consiste en clasificar a los pacientes en función de su riesgo de progresión y de su probabilidad de respuesta48. Del mismo modo que empleamos las herramientas genéticas, como los haplotipos HLA, DR y el estudio a gran escala de polimorfismos (HapMap), en el futuro inmediato debemos transformar nuestros conocimientos sobre la epigenética de la AR en herramientas con valor predictivo o pronóstico, e integrarlas a las ya disponibles. De forma simplificada, algunas de las medidas podrían ser la determinación del ratio HAT/HDAC y el grado de metilación del ADN en las muestras relevantes de los pacientes. En la actualidad ya se está trabajando en obtener estos patrones a gran escala, además de en el mapeo del sistema de microRNA (MirMap), no mencionado en esta revisión, pero de igual trascendencia para comprender la patogenia de las enfermedades autoinmunes49. El conocimiento de los mecanismos epigenéticos nos está proporcionando los medios para conocer mejor y utilizar mejor los tratamientos ya existentes para la artritis reumatoide. Asimismo, algunos circuitos patogénicos aparecen como candidatos ideales a ser reprogramados en los fibroblastos sinoviales y en las células mononucleares de los pacientes. De momento, para el uso de los agentes epigenéticos en la AR deberá optimizarse su selectividad y su captación por el tejido diana, con objeto de evitar la aparición de efectos indeseables. No obstante, en analogía con lo observado con los tratamientos biológicos, el éxito de las terapias epigenéticas se basará en la correcta selección de los pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la Fundación Conchita Rábago y a la Fundación de la Sociedad Española de Reumatología, por las ayudas recibidas para ampliación de formación durante 2006–2007.


