



Pese a los avances terapéuticos en las enfermedades autoinmunes e inflamatorias, muchos pacientes no logran un control adecuado de la enfermedad. De ahí la necesidad de optimizar el uso de las terapias biológicas y de explorar nuevas opciones terapéuticas. La disponibilidad de fármacos que inhiben proteínas-cinasas ya es una realidad en especialidades como oncología y hematología, donde los resultados asociados a la evolución clínica de la enfermedad han sido prometedores. La principal ventaja de estos fármacos es la administración oral, que podría favorecer la adherencia del paciente y reducir los costes asociados al tratamiento. Tofacitinib, inhibidor de tirosinas-cinasas, actualmente es el único fármaco de esta categoría aprobado para el tratamiento de la artritis reumatoide por la FDA. Estas dianas terapéuticas son evaluadas actualmente en diversas enfermedades autoinmunes e inflamatorias. Sin embargo, el conocimiento y la comprensión de las vías de señalización intracelular siguen siendo limitados, persistiendo dudas en cuanto al mecanismo de acción, la eficacia y los posibles efectos secundarios asociados al uso de estos nuevos fármacos.
Although advances in biological medicine have seen significant progress in the treatment of autoimmune and inflammatory disease, many patients do not experience a satisfactory response. Hence, there are two challenges facing the medical research community. The first is to continue development in the field of existing biological therapies, such as monoclonal antibodies. The second is to open new frontiers of research and explore treatment alternatives for non-responders to other therapies. Attention has increasingly turned to the therapeutic potential of small molecule weight kinase inhibitors (SMKIs), currently used extensively in oncology and haematology. Initial research into the therapeutic value of SMKIs for autoimmune and inflammatory diseases has been encouraging. SMKIs are taken orally, which reduces cost for the health provider, and could increase compliance for the patient. This is why research is now focusing increasingly on SMKIs as a new generation line of treatment in these diseases. Tofacitinib, an inhibitor of Janus-kinase, is currently the only drug approved for the treatment of rheumatoid arthritis by FDA. However, much more needs to be done to understand the intracellular signalling pathways and how these might affect disease progression before solid conclusions can be drawn.
Hasta hace un poco más de 10años las opciones terapéuticas en el tratamiento de patologías como artritis reumatoide (AR), lupus eritematoso sistémico (LES), espondiloartropatías (EA), psoriasis (Pso) y enfermedad intestinal inflamatoria (EII) como la enfermedad de Crohn (EC) y la colitis ulcerosa (CU) eran muy limitadas. Actualmente existe un amplio espectro de opciones terapéuticas que incluye anticuerpos monoclonales dirigidos contra interleucinas (IL), receptores de IL, receptores para el reconocimiento antigénico y comunicación intercelular1. Recientemente las proteínas-cinasas (PC) han emergido como dianas terapéuticas para el tratamiento de estas enfermedades, lo que ha conducido al desarrollo de inhibidores que bloquean la actividad de estas proteínas2. Todo esto ha contribuido a profundizar el conocimiento de los mecanismos inmunológicos subyacentes a la enfermedad y la respuesta terapéutica.
Proteínas-cinasas. Definición y función en relación con autoinmunidad e inflamaciónLas PC son enzimas que modifican bioquímicamente otras proteínas y/o enzimas, activándolas o desactivándolas, dependiendo del objetivo de la comunicación intracelular desde la membrana hacia el núcleo. A partir de la secuenciación del genoma humano y los avances en proteómica se han identificado más de quinientas PC, las cuales han sido catalogadas dentro de un sistema denominado «cinoma humano»3. Las PC han sido clasificadas dependiendo de su localización celular y función en 7 grupos principales, dentro de los cuales se encuentran las tirosinas-cinasas (TC). Las TC son enzimas que sirven de mediadoras entre la recepción de una señal extracelular y la ejecución de una respuesta efectora. La activación de las TC se produce mediante la fosforilación o transferencia de grupos fosfatos al grupo hidroxilo de los residuos de tirosina de la enzima4.
Las PC exhiben su actividad catalítica con: a)redundancia: la consecución de una acción o respuesta puede darse mediante la activación simultánea de diferentes enzimas no relacionadas entre sí; b)pleiotropismo: la activación de una enzima específica puede favorecer diferentes respuestas celulares, y c)sinergia: la activación de varias vías de señalización es necesaria para conseguir un efecto específico, y así el correcto funcionamiento de la comunicación y el funcionamiento celular.
Ahora bien ¿cuál es el papel de las PC en el marco del sistema inmunológico? Pues el complejo funcionamiento de las células del sistema inmune depende en parte de las PC por su papel determinante en la señalización de los procesos celulares de crecimiento, maduración, diferenciación, migración, inflamación, envejecimiento y apoptosis. Por ello es fundamental que la activación de las PC sea eficaz, específica y correcta, ya que defectos en la activación y regulación de estos mecanismos pueden afectar el sistema endocrino y/o favorecer el desarrollo de fenotipos celulares malignos y/o anómalos, como en el caso del cáncer y la autoinmunidad2,5,6.
¿En qué consiste el proceso de señalización intracelular?Como hemos mencionado anteriormente, el objetivo de la señalización intracelular es la elaboración de una respuesta nuclear a partir de la recepción de un estímulo que es reconocido por los receptores de membrana celular. Las PC pueden formar parte de estos receptores o estar asociadas a la membrana celular, siendo los receptores de 2 tipos: receptores con actividad cinasa intrínseca y receptores sin actividad cinasa intrínseca pero con la asociación de las cinasas en su región citoplasmática7.
Este proceso de señalización se puede resumir de la siguiente manera:
- a)
Reconocimiento de citocinas como quimiocinas, interleucinas, factores de crecimiento o antígenos por parte de receptores específicos que se encuentran sobre la membrana citoplasmática.
- b)
Unión de este receptor al estímulo que induce cambios conformacionales del receptor y proteínas adyacentes.
- c)
Estos cambios conformacionales favorecen la activación de las cinasas, y en el caso de las TC, la fosforilación de sus residuos de tirosina.
- d)
La activación de las cinasas desencadena la activación de otras proteínas a manera de cascada, en dirección hacia el núcleo celular.
- e)
Inicio de la transcripción génica para la síntesis de proteínas, o la iniciación de procesos celulares como la diferenciación, maduración, proliferación o apoptosis celular. También se puede desencadenar la síntesis/activación de proteínas reguladoras y/o inhibidoras como las fosforilasas, que en el caso de las TC interrumpen el proceso de señalización intracelular por defosforilación (fig. 1).

La red de comunicación intracelular está compuesta por diversas «vías de señalización», donde las PC participan según el objetivo de la señalización y la subpoblación celular en la que se encuentran. Para comprender y describir el proceso o cascada de señalización se utilizan los términos «up-stream» y «down-stream». La traducción literal al castellano de estos 2 términos sería «corriente arriba» y «corriente abajo», analogía del flujo de una corriente de agua que es utilizada para indicar la dirección y la posición de las proteínas dentro de la cascada de señalización intracelular. Por lo tanto, cuando se habla de que una acción sucede up-stream significa que ocurre en dirección a la membrana celular como el reconocimiento y la unión de los receptores celulares. Por otro lado, cuando se dice que esta acción sucede down-stream significa que ocurre en dirección al núcleo celular como la transcripción génica. Por consiguiente, si hablamos de una TC como la Janus kinase 3 (JAK-3), todo aquello que la activa y/o estimula, como las citocinas que se unen al receptor de la cadena γ-común, está up-stream (por encima de ella en la cascada de señalización) y todo aquello que la enzima en sí misma activa y/o regula, como por ejemplo la activación y el desarrollo del linfocitoT y natural killer (NK) y su homeostasis, está down-stream (por debajo de ella en la cascada de señalización). En la tabla 1 se describe detalladamente el significado de estos términos y de las siglas de cada una de las proteínas, genes y factores de transcripción descritos en esta revisión.
Abreviaturas
| ADN | deoxyribonucleic acid (DNA) | ácido desoxirribonucleico |
| AMA | American Medical Association | Sociedad Médica Americana |
| APhA | American Pharmacists Association | Sociedad Americana de Farmacéuticos |
| AR | rheumatoid arthritis (RA) | artritis reumatoide |
| ATF-2 | transcription factor 2 response to stress and DNA damage | factor de transcripción 2 que responde al estrés y al daño del ADN |
| ATP | adenosine triphosphate | adenosina trifosfato |
| BCR | B cell receptor | receptor del linfocito B |
| Bcl-6 | B-cell lymphoma 6 protein | proteína 6 del linfoma del linfocito B |
| Bcl-xl | B-cell lymphoma-extra large | proteína del linfoma del linfocito B gigantes |
| Bfl-1 | BCL2 protein family from a human fetal liver | familia de proteínas BCL2 del hígado fetal humano |
| Bmx | bone marrow tyrosine kinase gene in chromosome X protein | gen en el cromosoma X de la TC de la medula ósea |
| Btk | Bruton's tyrosine kinase or BPK (B cell progenitor kinase) | tirosina-cinasa de Bruton o cinasa del linfocito B progenitor |
| CD95 | apoptosis antigen 1 or Fas receptor | antígeno apoptótico 1 o receptor de FasL |
| CHMP | Committee for medicinal products for human use | Comité de medicamentos para uso humano |
| CU | ulcerative colitis (UC) | colitis ulcerosa |
| COX | cyclooxygenase | ciclooxigenasa |
| CREB | cAMP response element-binding protein | proteína de unión a la respuesta de cAMP |
| down-stream | down-stream | señalización hacia abajo en la cascada |
| DUSP | dual specificity protein phosphatase gene | genes que codifican proteínas-fosfatasas que inactivan p38, JNK y ERK |
| EA | ankylosing spondylitis (AS) | espondilitis anquilosante |
| EC | Crohn's disease (CD) | enfermedad de Crohn |
| Egr-1 | early growth response protein 1 | proteína de respuesta temprana al crecimiento 1 |
| EII | inflammatory bowel disease (IBD) | enfermedad intestinal inflamatoria |
| Elk-1 | E-26-like protein 1 | proteína tipo E-26 |
| EMA | European Medicines Agency | Agencia Europea del Medicamento |
| Emt | epithelial-to-mesenchymal transition | transición del epitelio mesénquimal |
| ERK | extracellular signal-regulated kinase | PC que regula señales extracelulares |
| Etk | endothelial and epithelial tyrosine kinase | TC endotelial y epitelial |
| FasL | Fas ligand or CD95 ligand | ligando del receptor de Fas o CD95 |
| FDA | Food and Drug Administration | Agencia reguladora de alimentos y medicamentos |
| GM-CSF | granulocyte macrophage colony-stimulating factor | factor estimulante de colonias de granulocitos y monocitos |
| IL | interleukin | interleucina |
| INN | International Nonproprietary Name | Programa internacional de nombres genéricos de la Organización Mundial de la Salud (OMS) |
| IFN | interferon | interferón |
| ITAM | immunoreceptor tyrosine-based activation motif | inmunoreceptores activadores que contienen tirosina |
| ITK | IL-2 Inducible T-cell Kinase | cinasa del linfocito T |
| JNK | c-Jun N-terminal kinase | PC que fosforila la proteína c-Jun |
| LES | systemic lupus erythematosus (SLE) | lupus eritematoso sistémico |
| LPS | lipopolysaccharide | lipopolisacáridos |
| Lyn | Lck/Yes related novel tyrosine kinase | TC asociadas Lck y Yes |
| MAPK | mitogen activated protein kinase | PC activada por mitogenos |
| MAPKK | MAPK: mitogen activated protein kinase | PC activada por mitogenos que activan MAPK |
| MAPKKK | MAPKK: mitogen activated protein kinase | PC activada por mitogenos que activan MAPKK |
| MEF2C | myocyte enhancer factor 2 | factor potenciador de miocitos 2 |
| MMP | matrix metalloproteinase | metaloproteinasas |
| MSK-1/2 | mitogen- and stress-activated kinase 1 and 2 | PC 1 y 2 activada por mitógenos y el estrés oxidativo |
| NFAT | nuclear factor of activated T-cells | factor nuclear que regula la activación del linfocito T |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells | factor nuclear que regula la transcripción de las cadenas ligeras kappa (κ) |
| NK | natural killer cells | linfocitos asesinos naturales |
| Oct-2 | transcription factor 2 | factor de transcripción 2 |
| Pax-6 | paired box protein 6 | proteína pareada 6 |
| PC | protein kinase (PK) | proteína cinasa |
| PIAS | protein inhibitor of activated STATs | proteína inhibidora de la transcripción de STAT |
| PIP 2 y 3 | phosphatidylinositol bi and trisphosphate | fosfatidil inositol bi y trifosfato |
| PI3K | phosphatidylinositol 3 kinase | PC de fosfatidil inositol 3 |
| PKC | protein kinase C | proteína-cinasa C |
| PLC | phospholipase C | fosfolipasa C |
| Pso | Psoriasis | psoriasis |
| PTEN | phosphatase and tensin homolog | fosfatidilinositol-3,4,5-trisfosfato 3-fosfatasa |
| PTI | immune thrombocytopenic purpura (ITP) | púrpura trombocitopénica autoinmune |
| PTP | protein tyrosine phosphatases | tirosinas-fosfatasas |
| RANKL | receptor activator of NF-kB ligand | ligando del receptor activador del NF-kB |
| SHP-1 | Src homology region 2 domain-containing phosphatase-1 | fosfatasa 1 de las tirosinas-cinasas Src |
| RHOA | ras homolog family member A | proteína GTPasa que regula la actina |
| SMKI | small molecular weight kinase inhibitors | pequeñas moléculas inhibidoras de PC |
| SNP | single nucleotide polymorphism | polimorfismo de solo un nucleótido |
| SOCS | suppressors of cytokine signaling | supresor de la señalización de citocinas |
| Src | proto-oncogene c-Src | PC que fosforila el protooncogen SRC |
| STAT | signal transducer and activator of transcription | transductor de señal y activador de la transcripción |
| Syk | spleen tyrosine kinase | tirosina-cinasa del bazo |
| TC | tyrosine kinase (TK) | tirosina cinasa |
| TCR | T cell receptor | receptor del linfocito T |
| Tec | Tyr protein-kinases enzymes cytosolic | TC citosólica |
| TLR | Toll-like receptor | receptor tipo Toll |
| TNFα | tumor necrosis factor alpha | factor de necrosis tumoral alfa |
| Tsk | T-cell signaling protein | proteína señalizadora del linfocito T |
| up-stream | up-stream | señalización arriba en la cascada de señalización |
| USAN | United States Adopted Names | Comisión americana encargada de asignar los nombres genéricos a nuevos fármacos |
| USP | United States Pharmacopeial Convention | Convención farmacopea de Estados Unidos de América |
| UV | ultraviolet light | radiación ultravioleta |
| ZAP70 | zeta-chain-associated protein kinase 70kDa | proteína asociada a la cadena zeta de 70 kilo-dalton |
Existen 3 grandes subfamilias de mitogen activated protein kinase (MAPK): p38, extracellular signal-regulated kinase (ERK) y c-Jun N-terminal kinase (JNK). La p38 es una cinasa serina-treonina, y es la subfamilia hasta ahora mejor caracterizada: tiene 4 isoformas homólogas, denominadas alfa (α), beta (β), gamma (γ) y delta (δ), las cuales son productos de diferentes genes que catalizan una misma reacción. Las isoformas α y β son ubicuas, mientras que la γ se encuentra en músculo esquelético y la δ en páncreas, intestino delgado y testículo. Por otro lado, ERK y JNK tienen 2 y 3 isoformas, respectivamente8,9.
La p38 y la JNK son activadas up-stream por diversos estímulos, como Fas ligand (FasL), factores de crecimiento, citocinas proinflamatorias, lipopolisacáridos (LPS), proteínas virales y estrés celular osmótico en células dendríticas, neutrófilos, macrófagos y linfocitosT y B. Estas enzimas regulan down-stream procesos celulares fundamentales, como la regulación del ciclo celular, apoptosis, envejecimiento celular y la producción de citocinas como la IL-10. ERK es activada up-stream por agentes infecciosos, señales mitóticas, factores de crecimiento, hormonas y citocinas proinflamatorias, activando down-stream la síntesis de proteínas del citoesqueleto, la proliferación y la diferenciación celular8,10.
Esta vía es regulada negativamente por las cinasas MAPKK —MKK3 y MKK6— (MAPK-3 y 6) así como por tirosinas-fosfatasas (PTP) de MAPK como las llamadas dual specificity protein phosphatase (DUSP). Del mismo modo, la producción de IL1-RA e IL-10 limitan la producción de citocinas proinflamatorias y prostaglandinas señalizadas por esta vía11.
ERK, JNK y p38 se encuentran activadas en el tejido sinovial de pacientes con AR, demostrando que cumplen un papel importante en inflamación y daño tisular en enfermedades de índole autoinmune. Fallos de la regulación en estas vías de señalización inducen alteraciones en la respuesta inmune innata y adaptativa, proliferación de células tumorales, resistencia a la insulina, desarrollo de enfermedades neurológicas y/o degenerativas, fallos en el control de infecciones y autoinmunidad8,10. La p38 es una proteína clave en la regulación de la respuesta proinflamatoria, por lo que ha sido una de las primeras PC investigadas como diana terapéutica en autoinmunidad e inflamación12.
Vía «Syk-Btk»La familia Syk consta de 2 miembros: zeta-chain-associated protein kinase 70 (ZAP70) y spleen tyrosine kinase (Syk)13. ZAP70 restringe su expresión a linfocitosT y NK; por el contrario, Syk se expresa en todas las células hematopoyéticas, mastocitos y sinoviocitos. Syk se une a secuencias cortas de aminoácidos en la región citoplasmática de inmunorreceptores que contienen tirosina (immunoreceptor tyrosine-based activation motif [ITAM]) como los receptores Fcγ de macrófagos, neutrófilos, mastocitos, y los receptores de linfocitosT y B: T-cell receptor/B-cell receptor (TCR/BCR)14. En AR se ha observado que Syk es activado en los sinoviocitos por citocinas proinflamatorias como TNF e IL-1, los cuales inducen up-stream la activación de JNK, la expresión de IL-6 y metaloproteinasas (MMP)15. Del mismo modo, la activación de Syk favorece la síntesis de IL-12 e IL-13, así como los procesos de proliferación, diferenciación, supervivencia, degranulación y fagocitosis celular16.
La familia Btk está compuesta por 4 miembros: Bruton tyrosine-kinase/phosphoinositide 3-kinase (Btk/PI3K), IL-2 inducible T-cell kinase/epithelial-to-mesenchymal transition/tyrosine-protein kinase (Itk/Emt/Tsk), bone marrow tyrosine kinase gene in chromosome X protein/endothelial/epithelial tyrosine kinase (Bmx/Etk) y Tyr protein-kinase cytosolic enzymes (Tec)17. La Btk es una TC que se expresa en todas las células hematopoyéticas y linfocitos excepto en linfocitos T y células plasmáticas maduras, es fundamental en la linfopoyesis y se encuentra más down-stream en esta cascada de señalización de Syk. La presentación antigénica al BCR, IL-6 y eritropoyetina activan up-stream enzimas de la familia proto-oncogene c-Src (Src), las cuales fosforilan a Syk y subsecuentemente a Btk. Su fosforilación inicia múltiples procesos celulares, como proliferación, supervivencia, migración, angiogénesis, presentación antigénica y producción de citocinas18.
PI3K es una subfamilia de lípido-cinasas agrupadas del i al iv. Pueden ser activadas por el eje Syk-Btk, por receptores de coestimulación tipo Toll (toll like receptors [TLR]) y moléculas de adhesión. Las PI3K del grupoi actúan sobre la phosphatidylinositol bisphosphate (PIP2) generando un segundo mensajero, el phosphatidylinositol trisphosphate (PIP3), el cual se une y facilita la fosforilación de Btk por medio de Syk y Lyn. Estas enzimas tienen un papel importante en la migración leucocitaria, la degranulación de mastocitos y la entrada de calcio a la célula19.
La activación down-stream de estas enzimas favorece la activación de otras vías que regulan la inflamación como MAPK (p38, ERK1/2), phosphoinositide 3-kinase (PI3K), phospholipaseC (PLC) y factores de transcripción como nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) y nuclear factor of activated T-cells (NFAT). NF-kB es un importante complejo proteico de factores de transcripción que están implicados en un gran número de procesos celulares indispensables para la supervivencia y el funcionamiento celular, y NFAT regula la activación, la diferenciación y la maduración del linfocitoB20.
Tirosina-fosfatasas como Src homology region 2 domain-containing phosphatase (SHP)-1, SHP-2, protein kinase C (PKC) y phosphatase and tensin homolog (PTEN) regulan la actividad enzimática de Syk, Btk y PI3K en procesos celulares como apoptosis, adhesión, migración y proliferación. Mutaciones en el gen Btk, como la agammaglobulinemia ligada al cromosomaX (XLA), pueden bloquear la diferenciación y la maduración de los linfocitosB, resultando disfuncionales para la presentación antigénica. De igual manera la activación constitutiva de esta enzima y los fallos en la señalización del eje BCR-Btk favorecen el desarrollo y la supervivencia de fenotipos aberrantes de linfocitosB. En modelos de lupus murino se ha observado que la deficiencia en PI3K disminuye la supervivencia de linfocitosT CD4+, la producción de autoanticuerpos, la producción de TNF-α, la proteinuria y la glomerulonefritis21. Fallos en la regulación de estas enzimas pueden inducir enfermedades mediadas por anticuerpos y de tipo alérgico, como asma y rinitis alérgica, e inducir autoinmunidad por defectos en la tolerancia y en la presentación antigénica del linfocitoB, favoreciendo el desarrollo de enfermedades como la AR, la leucemia y el cáncer22-24.
Vía «JAK-STAT»Hasta la fecha se han identificado 4 miembros de la familia JAK: JAK-1, JAK-2, JAK-3 y TYK-2, los cuales se expresan ubicuamente en todas las células, excepto JAK-3, que está restringido a células hematopoyéticas. Estas TC son estimuladas up-stream por receptores de citocinas tipoi y ii. JAK-1 es activada por citocinas que se unen a receptores que contienen la cadena γ-común y la glucoproteína transmembrana 130; JAK-2, por receptores que contienen la glucoproteína transmembrana 130, IL-3 e interferón (IFN)-γ; JAK-3, por receptores que contienen la cadena γ-común, y por último TYK-2 es estimulada por IL-12 y LPS25. Estas enzimas se asocian estructuralmente a la región citoplasmática de los receptores de citocinas en forma de dímeros o trímeros. Cada combinación de JAK y/o TYK es modulada por estímulos específicos y ejerce una función diferente sobre la señalización celular y, por consiguiente, sobre el sistema inmunológico. Los dimeros JAK-1/JAK-3, JAK-1/TYK-2, JAK-1/JAK-2 y JAK-2/TYK ejercen diferentes funciones down-stream propias de la inmunidad innata y adaptativa, mientras que otros, como el dímero JAK-2/JAK-2, regulan la maduración y la diferenciación de los linajes hematopoyéticos26-27.
La fosforilación de las JAK/TYK induce la fosforilación de los factores de la transcripción signal transducers and activators of transcription (STAT). Estos factores son una familia compuesta por 7 miembros: STAT1, 2, 3, 4, 5a, 5b y 6, que se encuentran más abajo en la cascada de señalización y se asocian como homodímeros o heterodímeros que, al ser fosforilados por JAK, se dimerizan y translocan al núcleo donde se unen a los genes diana, aumentando o reprimiendo la transcripción génica y la función celular28.
Mutaciones en estas enzimas o fallos en la señalización se han asociado con el desarrollo de trastornos mieloproliferativos, autoinmunes e inflamatorios29,30. Estudios de asociación genómica han descrito el papel de determinados genes de receptores, citocinas y enzimas asociados a cada uno de estos dímeros y/o trímeros JAK/TYK y la susceptibilidad a desarrollar alergia y diversas patologías autoinmunes e inflamatorias. Por ejemplo, se ha asociado la señalización del dímero JAK-1/TYK-2 por el IFN-α con Pso y EII; JAK-2/TYK-2 con IL-12 e IL-23 en enfermedad de Behçet, AR, LES, Pso, EII y EA; y el trímero JAK-1/JAK-2/TYK-2 con el estímulo de IL-6 y la susceptibilidad a desarrollar EII28.
Esta vía de señalización es regulada por diversas proteínas, como suppressors of cytokine signalling (SOCS), protein inhibitor of activated STAT (PIAS) y/o las tirosinas-fosfatasas SHP-1 y 2, que bloquean la unión de las STAT al ADN. Así mismo, esta vía de señalización regula y es regulada por otras vías de señalización, como PI3K, MAPK y NF-kB25,31.
Retos del desarrollo de inhibidores de tirosinas-cinasasLas enfermedades autoinmunes e inflamatorias son un grupo de enfermedades crónicas en las que, pese a la disponibilidad de múltiples opciones terapéuticas, un porcentaje de pacientes no responden de forma adecuada, probablemente debido a la complejidad del sistema inmunológico así como a las características propias de cada patología, del fármaco y del propio paciente. Por todo esto sigue siendo un reto seleccionar dianas terapéuticas apropiadas para el desarrollo de nuevos fármacos que aumenten las opciones terapéuticas cuando han fracasado las terapias convencionales y biológicas aprobadas actualmente. En las últimas décadas se han desarrollado diversos anticuerpos monoclonales dirigidos contra citocinas y receptores de citocinas propios del medio extracelular o localizados en la membrana celular. Por ello, y debido a que las JAK-STAT son enzimas pleiotrópicas que participan en la señalización intracelular de múltiples citocinas, se han desarrollado fármacos inhibidores de las TC como una nueva opción terapéutica más específica y efectiva en la inhibición de cada proceso patológico26.
Los inhibidores de las TC son pequeñas moléculas (small molecular weight kinase inhibitors [SMKI]) cuya principal ventaja es la administración oral frente a la vía subcutánea o intravenosa de los anticuerpos monoclonales. Esta vía de administración facilita la adherencia y la disposición del paciente a recibir el tratamiento y podría reducir el coste con respecto a los fármacos ya existentes. Este grupo de fármacos se absorben en estómago e intestino delgado. En el hígado, el sistema del citocromo P450 se encarga de su metabolismo, donde la presencia de polimorfismos genéticos (single nucleotide polimorfism [SNP]) puede alterar la farmacocinética, la biodisponibilidad y la eficacia del principio activo, como se ha descrito con otro tipo de fármacos metabolizados por este sistema. Los inhibidores de las TC bloquean el sitio de unión al adenosine triphosphate (ATP) de las TC. Este punto es crítico en el desarrollo de estos nuevos fármacos debido a que es indispensable que el bloqueo sea selectivo; para que la inhibición sea efectiva y específica, y para que no se produzca la inhibición de múltiples TC y aumente el riesgo de toxicidad y/o de efectos secundarios.
Este grupo de fármacos se ha incluido en la categoría de antineoplásicos y se les ha asignado la terminación con el sufijo-itinib (International Nonproprietary Name [INN])32. Del mismo modo, la nomenclatura utilizada para la asignación de nombres genéricos sigue las recomendaciones de un comité asesor de la Sociedad Médica Americana y la Sociedad Americana de Farmacéuticos (USAN, AM, USP, APhA)33.
Uso y perspectivas de los inhibidores de tirosinas-cinasasEl éxito clínico de varios de los inhibidores selectivos de TC en el tratamiento de tumores y leucemias (cáncer colorrectal, gástrico y mamario, carcinoma hepatocelular, sarcoma, melanoma metastásico, leucemia linfoblástica aguda y leucemia mieloide crónica) aprobados por la Food and Drug Administration (FDA) y la European Medicines Agency (EMA) ha favorecido el aumento de estudios preclínicos y clínicos, y de opciones terapéuticas para el tratamiento de enfermedades autoinmunes e inflamatorias (www.clinicaltrials.gov). De igual forma, ha permitido un mayor conocimiento del papel de las TC en la señalización intracelular y la inflamación34.
En modelos murinos de AR tratados con inhibidores de p38 (SB203580 y PH-797804), JNK (SP600125) y ERK (PD98059) se ha observado la disminución de factores proinflamatorios como prostaglandinas, IL-10 e inflamación articular35,36. Un estudio reciente describe que SP600125 inhibe la expresión de MMP, la pérdida de cartílago e incrementa el número de linfocitosT reguladores también en un modelo murino de AR37. Debido a la escasa eficacia observada con el uso de los inhibidores de JNK y ERK, existen solo estudios independientes de su uso en enfermedades autoinmunes. En EII el uso de inhibidores de p38 como semapimod (Ferring Pharmaceuticals) reduce la actividad inflamatoria en la EC, aunque sin respuesta del todo satisfactoria en sus formas graves38. El fármaco doramapimod (Boehringer Ingelheim) ha sido evaluado en AR, EA y EC, evidenciándose la disminución en la producción de citocinas proinflamatorias por parte de diferentes linajes celulares39,40. En AR, el pamapimod (Hoffman-La Roche) ha demostrado poca eficacia, así como una alta incidencia de infecciones y trastornos cutáneos41,42.
Con respecto a la inhibición de Syk, en estudios in vitro y modelos animales de AR, el PRT062070 (cerdulatinib), un inhibidor de Syk y JAK-1/3, se observó la disminución de la actividad inflamatoria y sinovitis por medio de la supresión de la producción de autoanticuerpos, por bloqueo de la señalización y activación del linfocitoB43. Un estudio con 2 modelos de ratón de AR tratados con P505-15, un inhibidor selectivo de Syk, corroboró su efectividad en el bloqueo de la señalización y activación del BCR y en la inhibición de la activación de los basófilos vía Fcγ44. En estudios clínicos, fostamatinib disodium (AstraZeneca-Rigel), un inhibidor de Syk, está siendo evaluado en pacientes con LES, Pso y púrpura trombocitopénica autoinmune. En modelos animales de AR, el profármaco R-788 de fostamatinib demostró la disminución en la producción de IL proinflamatorias, MMP y el retraso de la destrucción articular probablemente debido a su acción sobre linfocitosT y osteoclastos45. Por otro lado, se observó que prevenía el daño renal y la aparición de lesiones cutáneas en un modelo de lupus murino46. Sin embargo, a pesar de estos hallazgos, en un estudio preliminar en faseiii fostamatinib no fue superior al placebo en pacientes con AR y respuesta inadecuada a terapias biológicas47.
Diversos inhibidores de Btk, como dasatinib (Bristol-Myers Squibb), PCI-32765 (Ibrutinib, Janssen Research & Development, LLC), CC-292 (Azacitidine, Celgene Corporation) y GDC-0834 han sido aprobados para el tratamiento de mieloma múltiple, linfoma y leucemias de tipoB y están siendo evaluadas en estudios preclínicos y clínicos de AR y LES. Estudios in vitro han demostrado que la inhibición de Btk reduce la pérdida ósea en modelos murinos de osteoporosis inducida por receptor activator of NF-kB ligand (RANKL), así como la reducción en la producción de autoanticuerpos y el retraso en el desarrollo de patología renal en un modelo de lupus murino48-50. En un modelo de artritis inducida por colágeno en ratas el GDC-0834 favoreció la disminución de la inflamación articular24,51. Además, en un modelo de lupus murino se observó una disminución de depósitos de IgG, infiltrado y activación celular al tratar los ratones con el inhibidor RN-48652.
Los inhibidores de PI3K están siendo ampliamente estudiados en trastornos hematopoyéticos y tumores sólidos. En estudios preclínicos, moléculas como IC-87114 y CZC24832 disminuyeron la producción de IL-17, bloqueando la diferenciación de linfocitos Th17 y la generación de osteoclastos en modelos murinos de AR. Otro inhibidor de PI3K, el AS-605240, demostró reducción de inflamación, infiltrado de neutrófilos y erosiones. Esta misma molécula en un modelo de lupus murino incrementó la supervivencia y disminuyó la producción de autoanticuerpos y glomerulonefritis19.
Los resultados obtenidos con diversas moléculas dirigidas contra las enzimas JAK han sido prometedores en diversos ensayos clínicos para el tratamiento de Pso y AR, y la segunda generación de estas moléculas se caracteriza por ser más específicas y efectivas en la inhibición. En modelos de AR tratados con decernotinib (Vertex Pharmaceuticals Inc.), una molécula dirigida contra JAK-3, se observó una reducción de la inflamación articular53, aunque resultados clínicos preliminares revelaron un incremento del riesgo de infecciones y de los niveles de transaminasas54. ASP015K (Astellas Pharma Inc.), un inhibidor de JAK-3 y en menor medida de JAK-1 y JAK-2, ha favorecido la disminución del espesor de la epidermis y de la proliferación celular en pacientes con Pso55. Baricitinib (Incyte and Eli Lilly Company), un inhibidor de JAK-1/JAK-2, ha demostrado que reduce la actividad inflamatoria y favorece la preservación de cartílago y hueso en el modelo murino56. Ruxolitinib (Incyte Corporation y Novartis) es un fármaco oral que inhibe JAK-1/JAK-2; aprobado para el tratamiento de trastornos proliferativos, está siendo evaluado en su presentación tópica para el tratamiento en Pso. El estudio de esta molécula en modelos murinos favoreció la disminución en la fosforilación de STAT-3, y con ello la reducción del edema, la infiltración de linfocitos y las placas de psoriasis57. Resultados en estudios clínicos demostraron que este fármaco tiene buena tolerancia y eficacia en el tratamiento de la Pso58. Por otro lado, tofacitinib (Pfizer) es el único fármaco inhibidor de TC que ha sido comercializado en Estados Unidos y otros países para el tratamiento de la AR. Es un inhibidor funcional específico de JAK-3 que también inhibe JAK-1 y, en menor medida, JAK-259,60. En estudios preclínicos se observó que este inhibidor reducía los niveles de quimiocinas, interleucinas, reactantes de fase aguda y RANKL, favoreciendo la reducción de la reabsorción ósea mediada por los osteoclastos y la inflamación61. Del mismo modo, se ha observado que inhibe la respuesta proinflamatoria de linfocitos Th1 y Th1762 y previene el daño del cartílago en modelos animales63.
En estudios más recientes se ha observado que en pacientes con AR tratados con tofacitinib hay una inhibición de la proliferación de linfocitosT CD4 sin afectar el número absoluto de estos en la periferia. Además se ha identificado que un bajo número de linfocitosT CD8 antes de iniciar este tratamiento se correlaciona con el desarrollo de efectos secundarios durante el tratamiento en pacientes con AR64. Actualmente, dada la eficacia de tofacitinib en el tratamiento de la AR, se están realizando ensayos clínicos de faseiii en Pso, así como en CU, EC y EA.
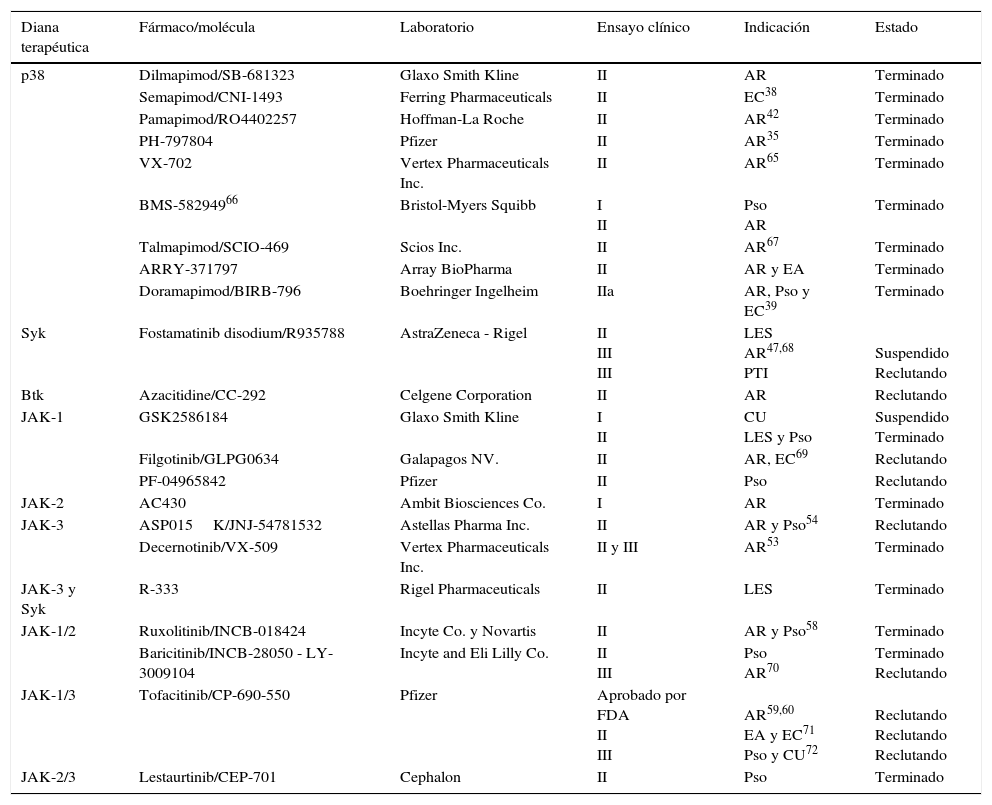
Un resumen de los principales ensayos clínicos desarrollados en las enfermedades autoinmunes e inflamatorias se detalla en la tabla 2. A pesar del reto que ha supuesto el desarrollo de estos inhibidores, comparado con los anticuerpos monoclonales, es decir, proteínas intracelulares vs. proteínas extracelulares, los efectos secundarios descritos parecen no ser diferentes a los observados hasta ahora con las terapias biológicas: neutropenia, altas tasas de infección, hepatotoxicidad y elevación de enzimas hepáticas, alteraciones de la función tiroidea, fatiga, hipertensión arterial, erupciones cutáneas, retraso en los procesos de cicatrización, mielosupresión, diarrea, elevación de la creatinina e hiperlipidemia, entre otros. Por ello, a día de hoy se está trabajando en el desarrollo de moléculas inhibidoras con mayor especificidad y con un efecto más dirigido a cada enfermedad.
Inhibidores de proteínas-cinasas evaluadas para el tratamiento de enfermedades autoinmunes e inflamatorias
| Diana terapéutica | Fármaco/molécula | Laboratorio | Ensayo clínico | Indicación | Estado |
|---|---|---|---|---|---|
| p38 | Dilmapimod/SB-681323 | Glaxo Smith Kline | II | AR | Terminado |
| Semapimod/CNI-1493 | Ferring Pharmaceuticals | II | EC38 | Terminado | |
| Pamapimod/RO4402257 | Hoffman-La Roche | II | AR42 | Terminado | |
| PH-797804 | Pfizer | II | AR35 | Terminado | |
| VX-702 | Vertex Pharmaceuticals Inc. | II | AR65 | Terminado | |
| BMS-58294966 | Bristol-Myers Squibb | I II | Pso AR | Terminado | |
| Talmapimod/SCIO-469 | Scios Inc. | II | AR67 | Terminado | |
| ARRY-371797 | Array BioPharma | II | AR y EA | Terminado | |
| Doramapimod/BIRB-796 | Boehringer Ingelheim | IIa | AR, Pso y EC39 | Terminado | |
| Syk | Fostamatinib disodium/R935788 | AstraZeneca - Rigel | II III III | LES AR47,68 PTI | Suspendido Reclutando |
| Btk | Azacitidine/CC-292 | Celgene Corporation | II | AR | Reclutando |
| JAK-1 | GSK2586184 | Glaxo Smith Kline | I II | CU LES y Pso | Suspendido Terminado |
| Filgotinib/GLPG0634 | Galapagos NV. | II | AR, EC69 | Reclutando | |
| PF-04965842 | Pfizer | II | Pso | Reclutando | |
| JAK-2 | AC430 | Ambit Biosciences Co. | I | AR | Terminado |
| JAK-3 | ASP015K/JNJ-54781532 | Astellas Pharma Inc. | II | AR y Pso54 | Reclutando |
| Decernotinib/VX-509 | Vertex Pharmaceuticals Inc. | II y III | AR53 | Terminado | |
| JAK-3 y Syk | R-333 | Rigel Pharmaceuticals | II | LES | Terminado |
| JAK-1/2 | Ruxolitinib/INCB-018424 | Incyte Co. y Novartis | II | AR y Pso58 | Terminado |
| Baricitinib/INCB-28050 - LY-3009104 | Incyte and Eli Lilly Co. | II III | Pso AR70 | Terminado Reclutando | |
| JAK-1/3 | Tofacitinib/CP-690-550 | Pfizer | Aprobado por FDA II III | AR59,60 EA y EC71 Pso y CU72 | Reclutando Reclutando Reclutando |
| JAK-2/3 | Lestaurtinib/CEP-701 | Cephalon | II | Pso | Terminado |
En noviembre del 2012 la FDA concedió la aprobación de tofacitinib (Xeljanz®) para el tratamiento de AR moderada-grave en adultos que hayan tenido una respuesta inadecuada o intolerancia a metotrexato (MTX). Según la ficha técnica, tofacitinib se administra por vía oral, en dosis de 5mg 2 veces al día, en monoterapia o en combinación con MTX u otros fármacos sintéticos, y no debe ser utilizado en combinación con terapias biológicas o inmunosupresores como azatioprina y ciclosporina. Actualmente, además de en Estados Unidos, tofacitinib está aprobado para el tratamiento de AR en varios países, incluyendo Japón, Rusia, Canadá y Suiza.
En la Comunidad Europea, en abril del año 2013 el comité de medicamentos para uso humano (Committee for Medicinal Products for Human Use [CHMP]) de la EMA consideró que, a pesar de la evidencia de reducción en los signos y síntomas de la AR y de la evidente mejora en la función física de estos pacientes, el beneficio no era suficiente respecto a los efectos secundarios asociados, como infecciones graves, perforaciones gastrointestinales y cáncer; a pesar de que el dossier presentado a la FDA fue el mismo presentado a la EMA. Así que, debido a la incertidumbre que generaba la magnitud de los riesgos y su gestión en la práctica clínica, se consideró que no compensaba su comercialización con respecto a los beneficios obtenidos con el tratamiento (EMA/CHMP/425279/2013, Procedure No. EMA/H/C/002542/0000). En vista de esto, la compañía solicitó una revaluación de los beneficios/riesgos del fármaco y el grupo de expertos, de conformidad con el artículo 12 del Reglamento CE N.° 726/2004, determino que la relación beneficio/riesgo del tofacitinib no era adecuada o no estaba suficientemente demostrada, así que denegó nuevamente la autorización para su comercialización (EMA/CHMP/425279/2013). Sin embargo, debido a que este fármaco lleva 2años comercializado en Estados Unidos, esperamos poder contar pronto con la evidencia de la práctica clínica, para corroborar los resultados obtenidos en los ensayos clínicos. Por otro lado, tofacitinib continúa siendo evaluado en diversos ensayos clínicos para su uso en otras indicaciones de índole autoinmune e inflamatoria, para posicionarse como una nueva opción terapéutica en el tratamiento de este grupo de patologías.
ConclusiónEl uso de los fármacos que inhiben PC como las TC podría cambiar en el enfoque terapéutico de enfermedades con fallos en la regulación del sistema inmunológico, debido a la ubicuidad de estas moléculas en el proceso de señalización intracelular y, por lo tanto, en su efecto en los procesos autoinmunes e inflamatorios. Esto podría beneficiar a aquellos pacientes que han fracasado a múltiples tratamientos por pérdida de eficacia y efectos secundarios. Así mismo, la introducción de estas moléculas de administración por vía oral cambiaría la adherencia del paciente y reduciría el coste asociado al tratamiento comparado con la administración de los anticuerpos monoclonales. Sin embargo, los resultados poco prometedores de algunos de estos inhibidores de PC nos han enseñado que no todas las vías de señalización intracelular son susceptibles de ser bloqueadas para obtener efectos clínicos beneficiosos. A medida que vayan obteniendo nuevos resultados en la práctica clínica sobre el uso de estas dianas terapéuticas, se podrá ir dilucidando qué vías de señalización intracelular son las más apropiadas de abordar para obtener un balance adecuado entre la eficacia clínica deseada y los efectos adversos. En este sentido y a la luz de las evidencias que se están obteniendo con el uso de los inhibidores de las JAK cinasas, como tofacitinib, la vía JAK-STAT se muestra como una de las principales dianas terapéutica para el desarrollo de nuevos fármacos y el tratamiento de enfermedades del sistema inmunológico.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLara Valor ha recibido honorarios profesionales por parte de: Abbvie, Bristol-Myers Squibb, Pfizer, Roche Farma, UCB, MSD.
Diana Hernández-Flórez declara no tener conflicto de intereses.
Las autoras agradecen a los Dres. Luis Carreño Pérez, Francisco Javier López Longo (Servicio de Reumatología, Hospital General Universitario Gregorio Marañón. Madrid) y Gustavo Centeno Soto (Servicio de Farmacología Clínica, Hospital Puerta de Hierro. Madrid) por la lectura crítica de este manuscrito.


