


El lupus eritematoso sistémico (LES)es una enfermedad autoinmune crónica que afecta múltiples sistemas. La mielopatía es uno de los 19 síndromes neuropsiquiátricos relacionados al LES, definidos por el Colegio Estadounidense de Reumatología. Aunque infrecuente, es una manifestación grave que cursa con déficit motor y sensitivo, y disfunción de los esfínteres. La fisiopatogenia no se conoce claramente, pero podría estar relacionada con trombosis arterial y/o vasculitis. El diagnóstico se basa en los hallazgos clínicos, los exámenes de laboratorio y el uso de la resonancia magnética con gadolinio. El tratamiento estándar es la combinación de ciclofosfamida y glucocorticoides por vía intravenosa. En casos refractarios se han utilizado otros tratamientos, como plasmaféresis o rituximab.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that affects multiple systems. Myelopathy is one of 19 neuropsychiatric syndromes related to SLE defined by the American College of Rheumatology. Although infrequent, it is a severe manifestation, leading to motor and sensory deficits, and sphincter dysfunction. The pathogenesis is not clearly known, but may be related to arterial thrombosis and vasculitis. Diagnosis is based on clinical findings, laboratory tests and the use of gadolinium-enhanced magnetic resonance imaging. The standard therapy is the combination of intravenous cyclophosphamide and glucocorticoids. In refractory disease, other treatments such as plasmapheresis or rituximab have been used.
La mielitis aguda (MA) es un cuadro inflamatorio de la médula espinal que se caracteriza por daño neuronal y axonal, provocando parálisis o paresia, déficit sensitivo y disfunción autonómica. La incidencia en la población general es de 1 a 4 casos por millón de habitantes por año1. De las múltiples causas de la MA, las enfermedades autoinmunes sistémicas constituyen una causa importante y, dentro de ellas, el lupus eritematoso sistémico (LES) es una de las más frecuentemente relacionadas2.
La mielopatía por LES forma parte de los 19 síndromes neuropsiquiátricos asociados definidos por el Colegio Estadounidense de Reumatología (ACR en inglés)3 (tabla 1). Teniendo en cuenta las manifestaciones neurológicas comunes como la cefalea, los trastornos del humor y las alteraciones cognitivas, la mielopatía es uno de los trastornos neuropsiquiátricos más infrecuentes (entre 1-2% de los pacientes)4. En la mayoría de los casos ocurre dentro de los primeros 5 años desde el comienzo de LES4 y, en casi la mitad de los casos, es la primera manifestación4,5, con un porcentaje de recurrencia entre el 18 y el 50%4–6.
Criterios diagnósticos de mielitis en LES
| Criterios diagnósticos de mielopatía en LES propuesto en el año 1999 por el ACR3 |
| Inicio rápido (días o horas) de los siguientes signos/síntomas |
| Debilidad bilateral de los miembros inferiores que puede o no incluir a los superiores (paraplejía o tetraplejía). Puede ser asimétrica |
| Alteración de la sensibilidad con nivel sensitivo que se corresponde con el compromiso motor, con o sin disfunción intestinal-vesical |
| Criterio de exclusión |
| Lesión compresiva de/en la médula espinal (p. ej., prolapso de disco) |
| Lesión de la «cola de caballo» |
La fisiopatogenia aún no se conoce del todo. Basado en hallazgos anatomopatológicos y serológicos, se postula que la vasculitis y la trombosis de los pequeños vasos serían los 2 mecanismos más importantes responsables del daño neuronal y axonal4,7–9. Dependiendo del tipo de compromiso medular (extenso o limitado), un mecanismo puede explicar mejor el cuadro que otro. En el caso de la mielitis transversa, el frecuente compromiso a nivel torácico4,8 (sector con los vasos de menor calibre de la vasculatura medular y por ende más vulnerables a la trombosis) y la presencia en suero de anticuerpos antifosfolípidos (aPL)4,8, indican que la trombosis cumpliría un papel patogénico preponderante4,8–10. Sin embargo, este mecanismo no explicaría la mielitis longitudinal de compromiso continuo6.
Diversos trabajos indican una importante asociación entre los aPL y la mielopatía en el lupus4,8,9, aunque la prevalencia de serología positiva no es mucho mayor que la de los pacientes sin compromiso medular4,9,10. El mecanismo de acción más probable es la trombosis. También se postula que los aPL ejercerían un efecto citotóxico directo, lo que se correlaciona con la presencia de bandas oligoclonales en los pacientes con aPL positivos6,9,10. Otro mecanismo sería a través de la denominada «cooperación entre anticuerpos»9: la isquemia induciría la síntesis de acuaporina 4 con el posterior desarrollo de mielitis lúpica asociada al espectro de neuromielitis óptica (NMO) mediado por anticuerpos IgG anti acuaporina-4 (AQP4-IgG) u otro tipo de anticuerpo9,10.
A pesar de todo lo mencionado, el papel de los aPL es controvertido9,10. En una revisión sistemática9, se compararon 2 grupos con serología de aPL positiva y negativa, respectivamente, no hallándose diferencias en la tasa de recaídas y evolución clínica general. Paradójicamente, el compromiso torácico fue más frecuente en el grupo con aPL negativo. Otros datos a considerar es que en gran parte de los artículos publicados4–6,8,9, solo se han hecho determinaciones de anticuerpos anticardiolipinas (aCL) y/o anticoagulante lúpico sin expresar valor preciso o haciéndolo en unidades no estandarizadas y sin información de los valores de corte utilizados (para los criterios de clasificación del síndrome antifosfolipídico, el valor es de 40 UI por ELISA11). Tampoco hay información de los isotipos estudiados: por ejemplo, los anticuerpos aCL-IgM se pueden hallar positivos en múltiples procesos de manera inespecífica. Otra falencia de los trabajos es que, en general, los pacientes en estudio cuentan con una única determinación de aPL, no pudiéndose descartar un fenómeno transitorio11.
Si bien no se ha atribuido un papel patogénico concreto al anticuerpo anti-Ro/SSA, su asociación con mielitis recurrente es bien reconocida12. Se ha encontrado este anticuerpo en pacientes con MNO y en mielitis transversa aun sin cumplir diagnóstico de síndrome de Sjögren13. Curiosamente, en una de las series más grandes de mielitis lúpica9 se observó que los casos recurrentes tenían serología positiva para anti-Ro/SSA más frecuentemente que aquellos monofásicos (evento único).
Otro mecanismo fisiopatológico propuesto es la alteración de la barrera hematoencefálica por autoanticuerpos12, sobre todo en los casos de superposición con NMO, aunque no hay hallazgos consistentes que prueben esta hipótesis12.
La falta de captación de gadolinio en la resonancia magnética (RM) en algunos casos de mielitis, ha sugerido un mecanismo fisiopatológico del tipo hemodinámico: la inflamación medular, al producirse en un espacio anatómico rígido, generaría hipertensión venosa progresiva (debido a la compresión del plexo venoso dorsal) con la consiguiente disminución del gradiente de perfusión entre las arterias radiculares de la médula y el plexo venoso de la piamadre, generando isquemia medular9. Este mecanismo, aún no probado, no explicaría el proceso inflamatorio inicial.
Manifestaciones clínicasEn líneas generales, se manifiesta como un cuadro agudo, que progresa en horas o días, aunque gran parte de los casos tiene su nadir en las primeras 24 h9,14. Puede ser precedido por síntomas generales como fiebre, cefalea y vómitos6,9, y, luego de un corto periodo, comienza con parestesias y paresia de miembros inferiores, generalmente grave, pudiendo llevar a la paraplejía o, menos frecuentemente, a tetraplejía, déficit sensitivo y disfunción de esfínteres, que se expresa como incontinencia urinaria y fecal4–6,8,9.
El compromiso motor casi siempre es bilateral, aunque no necesariamente simétrico, y de gravedad variable, pudiendo ir desde la paresia leve a la tetraplejía4–6,8,14. El déficit motor más frecuente es la paraparesia espástica4,8.
El déficit sensitivo, al igual que el motor, es bilateral, con manifestaciones de distinta gravedad: desde la anestesia (por debajo del nivel de lesión medular) hasta una disociación termoalgésica exclusivamente4,6,8,15. El segmento más afectado es el torácico (de T5 a T8, especialmente T7)4–6,8 y, usualmente, está bien delimitado.
El compromiso del sistema autónomo es frecuente: incluye retención urinaria y parálisis intestinal que evoluciona a la incontinencia vesical y fecal4,11. Puede haber alteración vasomotora por debajo del nivel, con livideces y frialdad en los miembros.
La MA puede estar acompañada de otras manifestaciones neurológicas. La neuritis óptica (NO) se asocia de manera relativamente frecuente4,5,9 (20-50%). Otras menos comunes son la depresión, la alteración de la memoria, las convulsiones, la psicosis y la oftalmoplejía4,6.
Mielitis longitudinalDurante años se creyó que la mielitis longitudinal por LES era una forma infrecuente de presentación de este cuadro neurológico15,16. Sin embargo, en una revisión sistemática se halló que es la más frecuente16. Esto se debe, probablemente, a la mejoría en la calidad de la RM, que permitiría mejor visualización de lesiones medulares14. En la mayoría de los casos la extensión es mayor a 4 segmentos10,14, pudiendo visualizarse en la RM lesiones continuas o parcheadas15. Los segmentos cervicales y torácicos medios (T5-T8) son los que se observan afectados con mayor frecuencia6. Aunque normalmente se presenta en casos de LES con índices de actividad elevada, hasta un tercio ocurre con baja o nula actividad15.
Desde punto de vista clínico, las manifestaciones más frecuentes (80-90%) son el déficit sensorial y motor, y la disfunción de esfínter urinario. El grado de compromiso es variable: va desde el trastorno miccional leve hasta la anestesia de miembros inferiores, paraplejía o incluso tetraplejía. En el examen físico se halla, casi invariablemente, afectación sensitiva y alteración de los reflejos (hipo o arreflexia)6,15,16. Hasta un 30% presenta afectación en el tronco en forma de parálisis de pares craneales6. A su vez, en una revisión sistemática de mielitis por lupus y aPL, se halló que un 80% de los casos con NO asociada a mielitis lúpica presentaban compromiso extenso (mayor a 3 segmentos)10. En comparación con la mielitis transversa (compromiso menor a 4 segmentos), el compromiso neurológico es más grave y hay mayor evidencia de actividad inflamatoria sistémica (p. ej., en el líquido cefalorraquídeo [LCR]). Si bien el pronóstico es similar (solo el 14% tiene recuperación total), la disfunción sensitiva, aún después del tratamiento, es más frecuente6.
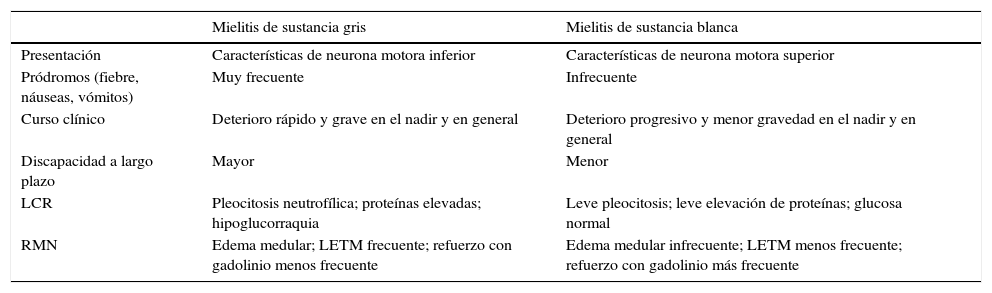
Compromiso de las sustancias blanca y grisUn trabajo8 que analizó a una cohorte de 22 pacientes con MA y LES reportó la presencia de 2 perfiles de compromiso medular bien diferenciados: un perfil con compromiso de sustancia gris medular, caracterizado por flaccidez e hiporreflexia, y otro que afecta a la sustancia blanca, que se manifiesta con espasticidad e hiperreflexia (tabla 2). El compromiso de la sustancia gris presenta un inicio hiperagudo, con mucha gravedad desde el principio (el nadir clínico se alcanza antes de las 6 h), pobre respuesta al tratamiento inmunosupresor y ocurre en contexto de altos índices de actividad de LES. Presenta síntomas prodrómicos como retención urinaria y fiebre, los cuales pueden ser tenidos en cuenta para el diagnóstico precoz. El compromiso de la sustancia blanca es de menor gravedad, con preservación de la fuerza, curso más indolente (no se alcanza al nadir clínico antes de las 72 h) y ocurre en el contexto de baja o nula actividad del LES. Curiosamente, en este trabajo se puede observar que entre los pacientes con perfil de compromiso de sustancia blanca existía un gran porcentaje (45%) que cumplía con criterios para NMO o presentaban anticuerpos anti NMO formando parte del espectro de NMO (81,8%).
Diferencias entre las mielitis de sustancia gris y de sustancia blanca en el LES
| Mielitis de sustancia gris | Mielitis de sustancia blanca | |
|---|---|---|
| Presentación | Características de neurona motora inferior | Características de neurona motora superior |
| Pródromos (fiebre, náuseas, vómitos) | Muy frecuente | Infrecuente |
| Curso clínico | Deterioro rápido y grave en el nadir y en general | Deterioro progresivo y menor gravedad en el nadir y en general |
| Discapacidad a largo plazo | Mayor | Menor |
| LCR | Pleocitosis neutrofílica; proteínas elevadas; hipoglucorraquia | Leve pleocitosis; leve elevación de proteínas; glucosa normal |
| RMN | Edema medular; LETM frecuente; refuerzo con gadolinio menos frecuente | Edema medular infrecuente; LETM menos frecuente; refuerzo con gadolinio más frecuente |
| Recurrencia | Muy rara | En más del 70% de los pacientes |
|---|---|---|
| Neuritis óptica previa | Ausente | Frecuente |
| NMO y/o NMO-IgG seropositivo | No | Frecuente |
| Actividad elevada de LES | Frecuente | Infrecuente |
LCR: líquido cefalorraquídeo; LES: lupus sistémico eritematoso; LETM: mielitis transversa longitudinalmente extensa; NMO: neuromielitis óptica; NMO-IgG: anticuerpo anti-acuaporina-4; RM: resonancia magnética.
La RM con gadolinio es considerada el método diagnóstico de elección para confirmar mielitis de cualquier causa1,2,17 incluyendo lupus4,8, como también para excluir otras causas de lesiones medulares no inflamatorias como compresión8. Si bien se pueden usar diferentes secuencias de RM para la detección de lesiones medulares, las secuencias por short-tau inversion recovery (STIR, en inglés) y T2 son las más sensibles para hallar lesiones medulares17. La recomendación actual es que la RM incluya toda la médula independientemente de la clínica1,2 y también el encéfalo a fin de descartar diagnósticos alternativos, como esclerosis múltiple (EM), siendo su asociación con LES muy infrecuente18. La tomografía computarizada no se recomienda para diagnóstico de mielitis por su falta de sensibilidad1,2 aunque tiene utilidad para descartar compresión como causa de mielopatía1,2.
El hallazgo más común es una lesión hiperintensa en T2, en general en la región central, acompañada o no de aumento del grosor medular, indicativo de edema. En la mayoría de los casos las lesiones refuerzan con contraste16. El número de focos se relaciona con la extensión del compromiso: en la mielitis transversa usualmente se visualiza un solo foco de aumento de señal en T25, mientras que en el compromiso longitudinal se pueden observar varios segmentos con aumento de señal, sobre todo cuando el compromiso es parcheado6. En los casos de atrofia medular, la RM evidencia atenuación de la señal4. Hay que tener en cuenta que un 30% de los casos puede no observarse lesiones al inicio4,15,16, visualizándose días después15. Obviamente, esto está relacionado con la potencia del resonador (números de Tesla) utilizado, por lo que se recomienda para aumentar el rédito que no sea menor de 1,5 Tesla1,2. Por esto, si la RM no reveló ninguna imagen, se debe repetir a los 2-7 días luego del comienzo del cuadro1. En los pacientes tratados, se puede observar reducción del número de imágenes hiperintensas, aunque no necesariamente se correlaciona con mejoría clínica14,15.
En un 30% de los casos se puede acompañar de lesiones en el cerebro y el tronco5,6, a veces indistinguibles de otras patologías desminelinizantes, como la EM, aunque algunos datos de la RM cerebral pueden ayudar al diagnóstico inicial. Por ejemplo, las lesiones subcorticales predominan en el síndrome antifosfolipídico y en LES, mientras que las lesiones periventriculares y del cuerpo calloso son más comunes en la esclerosis múltiple18.
Exámenes de laboratorioEn el examen de laboratorio se puede observar velocidad de sedimentación elevada, proteína C reactiva, leucopenia, linfopenia o anemia4,5. En el 95% los casos se acompañan de anticuerpos antinucleares (ANA) positivos al inicio del cuadro5,8 y en más del 50% de anticuerpos anti-ADN bicatenario (anti-ADNb)5,6,8 y/o hipocomplementemia6, lo que demuestra la importancia de estas determinaciones en los casos de comienzo de LES con mielopatía8. Como se mencionó anteriormente, existe una prevalencia del 50% de pacientes con aPL positivo en suero. Esta determinación es útil para el diagnóstico de LES en aquellos pacientes sin diagnóstico de LES previo o en los casos que se sospeche síndrome antifosfolipídico secundario (p. ej., infarto de médula).
El estudio del LCR es una herramienta fundamental para confirmar el diagnóstico de mielitis (de cualquier tipo) y para descartar infecciones1,17. Los hallazgos en el LCR pueden ser muy variables: van desde la normalidad (20-33%)4,5 hasta un líquido con patrón de meningitis bacteriana9 con pleocitosis, hipoglucorraquia y aumento de proteínas, aunque en el compromiso longitudinal se observa casi siempre pleocitosis a predominio de polimorfonucleares6. Cuando aún no está instaurado el tratamiento, la repetición de la punción muestra un LCR con mayor signo de inflamación6. También puede haber presencia de bandas oligoclonales, sobre todo en pacientes con aPL positivo en suero10.
La determinación del anticuerpo anti-NMO (tipo IgG) en suero es útil para distinguir a aquellos pacientes con mielopatía por LES asociado a NMO5,12,19, sobre todo en los casos de mielitis extensa5,12,19 o de compromiso de la sustancia blanca9. Se ha observado que la seropositividad para anti-NMO se mantiene a pesar de recibir inmunosupresión como glucocorticoides y/o ciclofosfamida9.
Diagnósticos diferencialesLa mielopatía por lupus se debe diferenciar de las causas compresivas (p. ej., tumores), infecciosas1,2,17, de patologías desmielinizantes como la EM18 o la NMO12,13,18,19 y de la mielitis idiopática1,2,14. En ocasiones, el diagnóstico diferencial al inicio es dificultoso ya que un gran porcentaje de los casos de LES comienzan con mielopatía, haciéndolo indistinguible de otras causas incluso con el estudio de laboratorio, siendo el diagnóstico retrospectivo en estos casos.
Neuromielitis óptica y mielopatía en lupusSi bien los criterios diagnósticos de ACR3 no incluyen la determinación de anticuerpos anti-NMO en la evaluación de los pacientes con mielopatía y lupus, varios autores proponen su realización por su implicancia en el pronóstico y la terapéutica5,12,13,19. No hay datos clínicos que puedan distinguir de manera taxativa ambas entidades: el compromiso medular puede ser indistinguible en los casos de compromiso de sustancia blanca o de mielitis longitudinal5,9,13,19. Actualmente, se postula que son 2 patologías distintas que pueden coexistir12,19 debido a factores genéticos y ambientales comunes que predisponen12. Sin embargo, en los casos de mielitis longitudinal recurrente sin diagnóstico previo de LES (sobre todo si se acompaña de NO), esta coexistencia no es tan clara: algunos interpretan este cuadro como NMO12,19 a pesar de presentar autoanticuerpos positivos como anticuerpos antinucleares o anti-Ro, otros lo definen como mielopatía por LES con compromiso de sustancia blanca5 y otros como una asociación de ambas entidades pero en etapas tempranas del desarrollo de LES13. Esto enfatiza la necesidad del trabajo multidisciplinario12,13 para llegar a un diagnóstico correcto.
TratamientoLa European League Against Rheumatism (EULAR, su sigla en inglés)20, en sus guías de tratamiento de manifestaciones neuropsiquiátricas en el lupus, recomienda la instauración temprana (idealmente dentro de las primeras horas de iniciado el cuadro) de metilprednisolona y ciclofosfamida por vía intravenosa (recomendación grado A). Aun en los casos que el LCR sugiera meningitis, se debe iniciar lo antes posible el tratamiento con glucocorticoides a dosis alta mientras se realizan los estudios microbiológicos. Sin embargo, debido a la baja prevalencia de mielitis en LES, existe poca evidencia sobre el tratamiento óptimo5,21. Existe un solo ensayo clínico aleatorizado21 de tratamiento del compromiso neurológico grave en el LES que demostró la utilidad de los glucocorticoides por vía intravenosa solos o asociados a ciclofosfamida, siendo más efectiva esta combinación22, aunque el subgrupo de pacientes con mielitis era muy pequeño para poder hallar diferencias significativas. Un reciente trabajo5 de mielitis asociada a LES evidenció que los pacientes que no reciben ciclofosfamida por vía intravenosa presentan un peor pronóstico neurológico. De este modo, se considera la combinación de glucocorticoides y ciclofosfamida por vía intravenosa como la terapia estándar5,20. Las dosis utilizadas en los trabajos publicados4–6,22 son: pulsos de 1 g de metilprednisolona por vía intravenosa por 3 días junto a ciclofosfamida por vía intravenosa a 0,75-1g/m2 de superficie corporal mensual por 6 meses a un año y luego cada 3 meses un año, junto con prednisona por vía oral 1mg/kg/día al cuarto día de comenzado el tratamiento, con un descenso gradual después de 1-3 meses. En los casos refractarios se puede adicionar plasmaféresis entre 6 a 10 sesiones4–9, aunque no parece mejorar el pronóstico4. También se ha utilizado gammaglobulina por vía intravenosa como terapia de inicio o en casos refractarios, sola o acompañando el tratamiento estándar, con respuesta variable5,6,9. La anticoagulación, si bien es usada en los casos de mielitis con aPL positivo4,6,8,16, no demostró que agregue ningún beneficio terapéutico a la inmunosupresión10.
Debido a la alta tasa de recurrencia, la EULAR recomienda un mantenimiento posterior con inmunosupresión, aunque menos intensa (nivel evidencia 2, recomendación grado D). Se han utilizado glucocorticoides a dosis bajas4,5,8,16,22, azatioprina5,6,8,16 y micofenolato de mofetilo5. La hidroxicloroquina disminuiría las recaídas5.
Durante más de 10 años se han publicado estudios que indican la utilidad del rituximab en cuadros graves o refractarios de lupus23, como las manifestaciones neuropsiquiátricas severas que no responden a los glucocorticoides y a la ciclofosfamida por vía intravenosa. Existen reportes de su uso en casos graves de mielitis (por falta de respuesta en la inducción o recaída frecuente), solo o en combinación con glucocorticoides, con una respuesta variable5,6. La dosis utilizada es de 375mg/m2 semanal por 4 semanas o 1.000mg separado por 2 semanas5,6,23,24. A pesar de su efectividad, más de un 50% recae al año23,24, por lo que se sugiere repetir un nuevo ciclo a los 6-12 meses23,24.
Además del tratamiento farmacológico, los pacientes requieren iniciar rehabilitación precozmente y otras medidas a fin de evitar las complicaciones por las secuelas (p. ej., úlceras por decúbito, infección urinaria por vejiga neurogénica)5.
Evolución y factores pronósticosEntre un 14-27% de los casos presentan resolución completa con tratamiento4–6,8. Los datos sobre recuperación parcial o ausencia de la misma publicados son muy variados (14-73% y 5-64%, respectivamente4–6,8). La recurrencia es frecuente: entre un 18 y un 50%4–6 va a presentar por lo menos un nuevo episodio dentro del primer año de ocurrido el cuadro aun con tratamiento óptimo4,6.
Existen diversos factores pronósticos a corto y largo plazo sobre la evolución del cuadro y el riesgo de recaída.
Clínicos: la gravedad del compromiso neurológico al inicio y la necesidad de cateterismo urinario son factores que predicen discapacidad severa a los 6 y 12 meses de ocurrido el cuadro, respectivamente5. El compromiso de la sustancia gris, a pesar de recibir inmunosupresión intensa, se asocia con peor pronóstico9.
Imagen: la evidencia de lesión en la RM4, el número de las mismas4,17 y su extensión6,16,17 son datos que están asociados a peor pronóstico en términos de respuesta a la medicación y discapacidad posterior. Sin embargo, un trabajo que comparó a pacientes con compromiso medular mayor a 4 segmentos y con afección menor no halló diferencia significativa en la respuesta al tratamiento, aunque el primer grupo persistió con mayor déficit sensitivo10.
Estudio de laboratorio: se ha visto que aquellos pacientes con LCR anormal tienen peor pronóstico neurológico10. La seropositividad de anti-NMO se asocia a un riesgo de recaída al año del 60%12.
Terapéuticos: la falta de adición de ciclofosfamida al tratamiento inicial es un factor de mal pronóstico neurológico, que predice discapacidad5. Así también, no usar hidroxicloroquina5 y el tratamiento de mantenimiento inadecuado (menor de 2 años y/o en dosis menores a las recomendadas) están asociados con recaídas dentro del año.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la doctora Carmen Lessa (jefa de la Unidad de Inmunología e Histocompatibilidad del Hospital Carlos Durand, Buenos Aires, Argentina).
A la doctora Mónica Perassolo (jefa del Servicio de Neurología del Hospital Carlos Durand, Buenos Aires, Argentina).
Al doctor José Luis di Pace (médico del Servicio de Neurología del Hospital Carlos Durand, Buenos Aires, Argentina)
A todos ellos gracias por vuestro apoyo y consejo para publicar este trabajo.


