



Mujer de 32 años de edad con antecedentes personales de hipotiroidismo en tratamiento sustitutivo, menstruante y sin hijos. Refería no tener antecedentes familiares de enfermedad osteoarticular. Relataba haber sido diagnosticada, en su país de origen, a la edad de 4 años de distrofia muscular. No se disponía de informes ni de historia previa en este sentido, refiriendo la paciente que dicho diagnóstico se había realizado por retraso en la adquisición de la edad de deambulación y que no se le había realizado biopsia muscular ni aportaba ningún estudio específico. Consultaba por artralgias mecánicas de largo tiempo de evolución, sin tumefacción articular, sin rigidez matinal y refería que desde hacía años los miembros superiores habían aumentado de tamaño y que el diámetro de muslos y piernas también habían aumentado. Por todo esto había acudido originalmente al Servicio de Neurología de nuestro hospital, donde tras la realización de analítica con CPK normal, electromiograma sin datos de miopatía y estudio de resonancia magnética de cintura pelviana sin datos de miositis, nos la remiten para estudio. A la exploración destacaba ausencia de sinovitis, recorridos articulares pasivos y activos limitados de forma global, fuerza muscular normal y aumento de longitud de miembros superiores. También llamaba la atención una marcha con cierta oscilación de la pelvis, lordosis lumbar exagerada y balanceo laterolateral. Los reflejos tendinosos eran normales y no había deformidad en los dedos de los pies ni de las manos. El estudio radiológico simple realizado mostró engrosamiento cortical y esclerosis de las diáfisis de los huesos tubulares con osteoesclerosis irregular y heterogénea con áreas moteadas de radiolucencia con mayor afectación endóstica que perióstica y estrechamiento de los canales medulares (fig. 1). Las epífisis estaban respetadas radiológicamente. Densitometría de cuello femoral Z-score +6,2 DE.
Discusión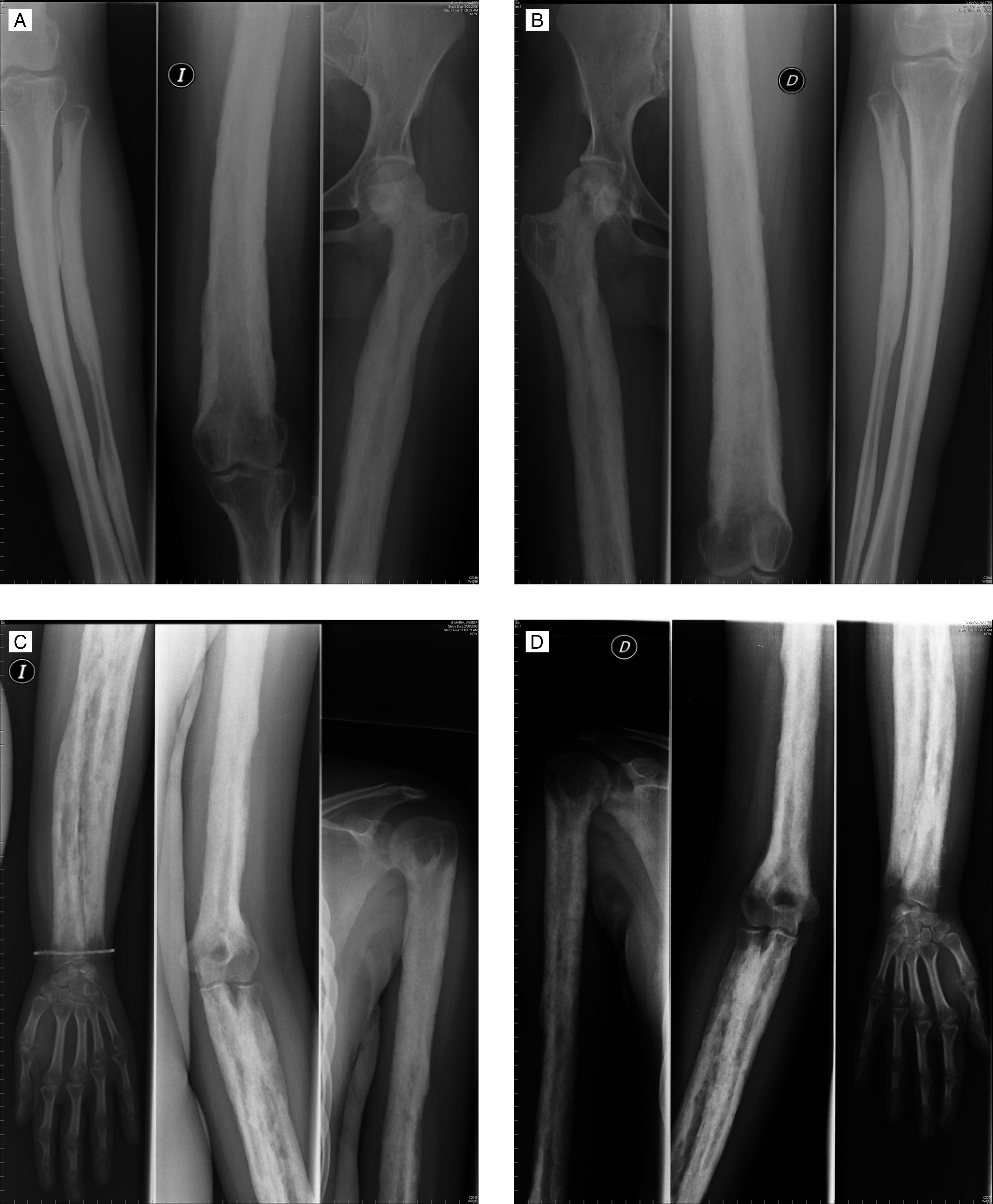
Los hallazgos radiológicos son sugestivos de displasia ósea. El número de displasias óseas es alto y no existe un consenso generalizado sobre su nomenclatura y clasificación; no obstante, con la expansión del conocimiento del genoma humano, para aquellas en las que el locus genético es conocido, ahora es posible la tipificación genética exacta. En este caso la afectación diafisaria con respeto de las epífisis hace pensar que se trata de una displasia del grupo hiperostosis craniotubular, siendo la más frecuente de las mismas la displasia conocida con el epónimo de enfermedad de Camurati-Engelmann. Se trata de una displasia con engrosamiento progresivo de las diáfisis de los huesos largos de forma bilateral y simétrica que a medida que avanza afecta a las metáfisis pero no a las epífisis1. El cráneo también puede estar afectado pero éste no era el caso de nuestra paciente. La presentación se suele dar en la infancia con dolor articular, debilidad muscular, marcha de pato y fatigabilidad. Su patrón de herencia es autosómica dominante y se debe a una mutación en el gen del TGFβ1 (transforming growth factor β1), de la cual se conocen 10 variantes2. El estudio genético en nuestra paciente confirmó la sospecha diagnóstica para la mutación c.652C→T, p.Arg218Cys, mutación ya descrita previamente para esta enfermedad2.
La paciente fue tratada con AINE según dolor articular, dado que la intensidad del dolor referido no parecía requerir mayor analgesia. Los fármacos más frecuentemente usados en la literatura han sido los glucocorticoides que, debido a su efecto antiinflamatorio, parecen aliviar el dolor óseo en estos pacientes. Los bifosfonatos, fundamentalmente pamidronato endovenoso, han sido utilizados para tratar esta enfermedad, pero con resultados contradictorios3,4.
DiagnósticoDisplasia ósea diafisaria tipo enfermedad de Camurati-Engelmann.


