





La amiloidosis secundaria puede encontrarse en algunas enfermedades autoinflamatorias monogénicas. Presentamos el caso de un varón de 83 años sin antecedentes patológicos de interés. Tras detectarse una anemia ferropénica, se realizó una gastroscopia y la biopsia duodenal evidenció amiloidosis secundaria de tipo AA. El estudio de enfermedades autoinflamatorias reveló la variante heterocigota p.R92Q en el gen TNFRSF1A, siendo negativas las pruebas complementarias para otras causas de amiloidosis. En el síndrome TRAPS la amiloidosis secundaria puede asociarse a mutaciones que afectan a residuos cisteína, no habiéndose evidenciado su asociación con la variante p.R92Q.
La amiloidosis secundaria puede estar presente en individuos portadores de la variante p.RQ92, por lo que es importante su diagnóstico para intentar prevenir posibles complicaciones.
Secondary amyloidosis can be found in some monogenic autoinflammatory diseases. In this study we present an 83-year-old man with no relevant medical history who presented with iron deficiency anaemia. In the study, a gastroscopy was performed with duodenum biopsy showing secondary AA-type amyloidosis.
Genetic analyses of monogenic autoinflammatory diseases revealed the heterozygous p.R92Q variant in the TNFRSF1A gene, with negative results in the complementary tests for other causes of amyloidosis.
In TRAPS, secondary amyloidosis has usually been associated with mutations affecting cysteine residues, but until now no association has been demonstrated with the p.RQ92 variant.
Secondary amyloidosis may be present in carriers of the p.RQ92 variant, therefore it is important to diagnose it to prevent possible complications.
El síndrome periódico asociado al receptor del TNF (TRAPS) es una enfermedad autosómica dominante caracterizada por episodios recurrentes de fiebre, mialgias migratorias, rash cutáneo, conjuntivitis, edema orbitario y dolor abdominal o torácico por inflamación muscular1,2, e incluso artritis crónica refractaria3. Puede aparecer leucocitosis, neutrofilia, trombocitosis y elevación de los reactantes de fase aguda. Se presenta el caso de un varón de 83 años que presentó amiloidosis de tipo AA y la variante p.R92Q en el gen TNFRSF1A.
Caso clínicoVarón de 83 años, con antecedentes de hemorragia digestiva y anemia ferropénica por gastritis y ulcus duodenal, hipertrofia benigna de próstata, hipertensión arterial en tratamiento con tamsulosina (0,4mg/24h), dutasterida (0,5mg/24h) y omeprazol (20mg/24h).
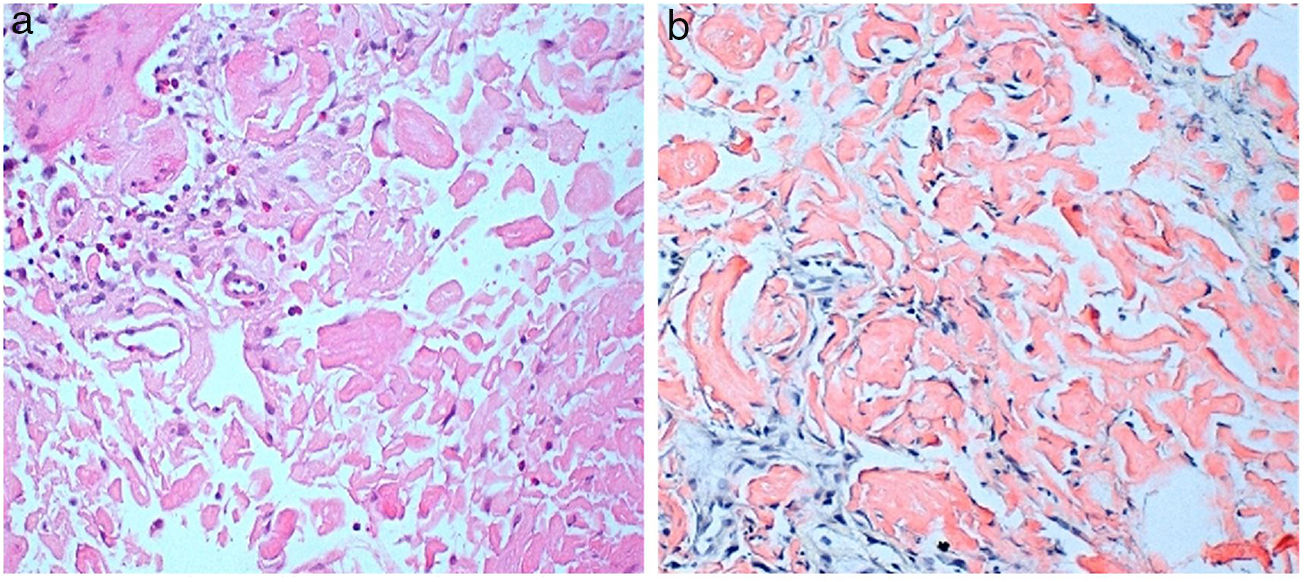
Consultó por cansancio e inestabilidad, siendo la hemoglobina de 7,3g/dl. La gastroscopia evidenció una úlcera duodenal. La biopsia gástrica mostró una gastritis crónica leve. En la biopsia duodenal se observó una mucosa focalmente ulcerada con vellosidades conservadas y una lámina propia expandida por un depósito de material amorfo eosinófilo (fig. 1A); dicho depósito se observaba también en las paredes capilares y en la submucosa, se acompañaba de un mínimo infiltrado inflamatorio linfocitario inespecífico; la tinción de Rojo Congo fue intensamente positiva y mostró birrefringencia verde manzana con luz polarizada (fig. 1B). También se practicó tinción inmunohistoquímica para la proteína AA, que fue intensamente positiva.
Se realizó una enteroscopia que demostró desde el bulbo distal, y de forma continua a lo largo del trayecto explorado, una mucosa intestinal con ulceraciones superficiales y de aspecto infiltrativo, con áreas con sangrado espontáneo al paso del endoscopio; los hallazgos más significativos se localizaron en el bulbo distal y la segunda y la tercera porción duodenales, con áreas de mucosa totalmente denudadas y ulceradas. La colonoscopia fue normal. La biopsia rectal no mostró material amiloide.
La TAC torácica era normal, mientras la TAC abdominal reveló un discreto engrosamiento con dilatación de la pared duodenal y primeras asas yeyunales (hasta 3,2cm) con afectación de la grasa adyacente, de probable origen inflamatorio; litiasis ureteral y vesical. La angio-TC abdominal no mostró hallazgos valorables.
Se realizó el estudio etiológico de la amiloidosis secundaria. Las funciones hepática y renal, el proteinograma, y la proteína amiloide A sérica fueron normales. Los anticuerpos antinucleares, el factor reumatoideo, el péptido citrulinado, los anticuerpos anticitoplasma de neutrófilo y la beta-2-microglobulina sérica fueron negativos. La proteína C reactiva (PCR) era normal al ingreso, pero se evidenciaron elevaciones intermitentes de alrededor de 50mg/l (normal 0,0-6,0), en alguno de los controles rutinarios, no presentando sintomatología asociada. La biopsia de grasa abdominal no evidenció material amiloide. La proteinuria de 24 h fue negativa.
El estudio de los genes para fiebre mediterránea familiar (MEFV) y la polineuropatía amiloide familiar (TTR) fueron negativos, mientras en el análisis del gen TNFRSF1A reveló la variante heterocigota p.R92Q (fig. 2). El electrocardiograma era normal y en el ecocardiograma se observó un ventrículo izquierdo no dilatado ni hipertrofiado, fracción de eyección conservada, con alteración de la relajación. En el electromiograma se apreciaron leves signos de afectación radicular crónica L4, L5 y S1 bilaterales, no había signos concluyentes de polineuropatía. La resonancia magnética lumbar-sacra mostró cambios osteodegenerativos en las articulaciones interapofisarias L4-L5-S1.
Discusión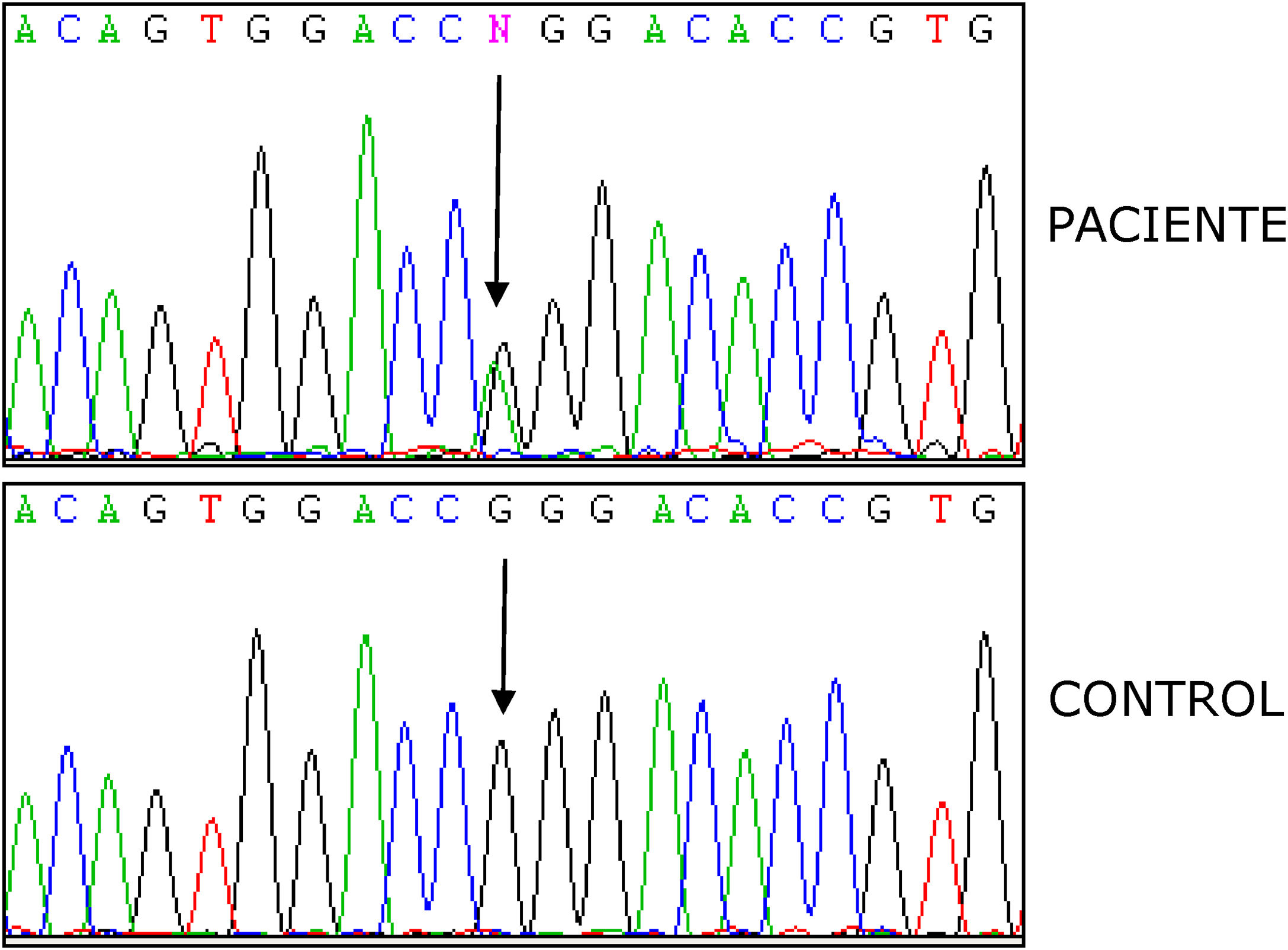
El síndrome TRAPS es una enfermedad autoinflamatoria monogénica que puede afectar a diferentes grupos étnicos. Las mutaciones causantes de este síndrome se localizan en el gen TNFRSF1A, habiéndose descrito alrededor de 114 variantes. Entre todas ellas, destaca por su frecuencia la variante p.R92Q, clasificada actualmente como variante de significado incierto, y presentando además una baja incidencia de amiloidosis secundaria4.
En el síndrome TRAPS los síntomas suelen aparecer durante la primera década de la vida. En el caso de inicio en edad adulta, es frecuente la identificación de variantes de baja penetrancia5. La manifestación más frecuente del síndrome son los episodios febriles prolongados (entre 1 y 4 semanas), que recurren a intervalos irregulares de semanas o meses, acompañados de mialgias y exantema cutáneo migratorio. Puede haber factores desencadenantes, como infecciones, traumatismos, estrés, etc.6.
La elevación prolongada de los reactantes de fase aguda, y muy especialmente la proteína sérica del amiloide, es la que incrementa el riesgo de amiloidosis de tipo AA. El paciente aquí descrito presentó PCR elevada en algunos de los análisis rutinarios sin presentar ningún síntoma. No se apreció hipertrofia ventricular concéntrica ni se evidenció el depósito de amiloide en las biopsias de la grasa abdominal ni rectal. La biopsia duodenal descartó la enfermedad de Crohn (EC), dado que en la EC puede haber ulceración y fisuras, pero con predominio de un infiltrado inflamatorio linfoplasmocelular moderado severo, granulomas e inflamación aguda glandular, no presentes en este caso.
La TAC torácica y abdominal, la gastroscopia, la enteroscopia y la colonoscopia no evidenciaron neoplasias, y el proteinograma no evidenció bandas monoclonales.
En los TRAPS puede aparecer amiloidosis7,8, pero hasta ahora no se había evidenciado su asociación con la variante p.RQ922.
Como conclusión, la amiloidosis puede estar presente en los portadores de la variante p.RQ92, por lo que es importante su diagnóstico para intentar evitar posibles complicaciones.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


